Ligand PPF-Ph2 was reported by Hayashi and Kumada in 1980 (synthesised from PPFA) and later by Togni in 1994. The Togni paper invoked the name ‘Josiphos’ for this type of ligand (named after a non-author technician in the group), and also reported ligands Josiphos-1 and Josiphos-3. The former is often just referred to as Josiphos. The ligand synthesis methodology provides considerable scope for varying the steric and electronic properties of the phosphorus groups, and as a result there are numerous Josiphos type ligands commercially available. The following lists reactions, organised by metal (Pd, Ru, Os, Rh, Ir, Cu, Co, Ni and organocatalysis), for which the application of these ligands has resulted in >80% ee. ACE = Asymmetric Catalytic Efficiency. Literature covered until end 2017 (essentially complete for Josiphos-1 but not necessarily for the other Josiphos derivatives – 132 entries).

Palladium
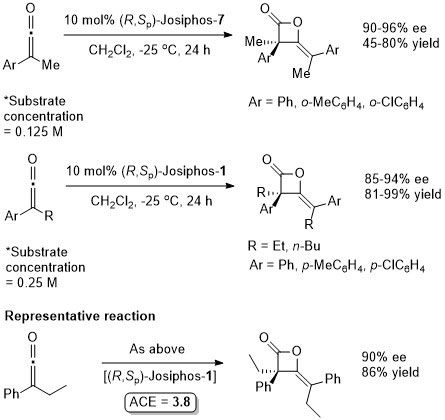
Pd – Allylic alkylation JACS94-116-4062

Orgmet04-23-2362. Reaction repeated under almost exactly the same conditions with (R,Sp)-Josiphos-1 to give S product 92% ee, 98% yield. JC08-254-79. For this reaction use of a heterogeneous catalyst derived from 5 wt% Pd/Al2O3 (1.4 mol% Pd) and R,Sp-Josiphos-1 (0.23 mol%) resulted in (S)-product of 88% ee (19% conversion) in THF at 60 oC for 24 h (substrate concentration = 0.18 M). Homogeneous Pd catalysis with R,Sp-Josiphos-1 in THF gave 92% ee (S). With 8 mol% (R,Sp)-Josiphos-1 and 2 mol% [Pd(η3-C3H5)Cl]2 in THF heated at reflux a 84% ee (S), 86% yield was obtained with CH2(CO2Me)2 (2 eq.) and ZnEt2 (2 eq.) (i.e. via zinc enolate) See Tet06-62-1756. A polymer-supported palladium source has also been used with (R,Sp)-Josiphos-1 (93% ee) – see JMCA03-201-131.
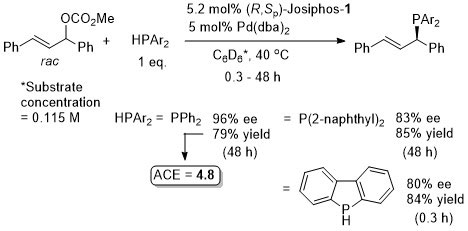
Pd – Allylic phosphination Angew08-47-4878
Pd – Allylic phosphonation EJOC14-6846
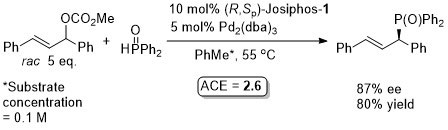
Absolute configuration not stated so assigned by comparison to JACS94-116-4062. For this reaction under the same conditions an ee of 97% (yield 95%) was achieved with (R)-BINAP. Reaction further exemplified using this ligand.
Pd – Hydrophosphorylation OL06-8-2099
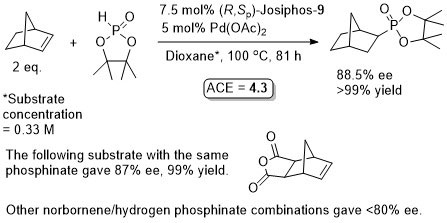
The absolute configuration of the products was not established. No mention of exo/endo so presumably completely selective for the former.
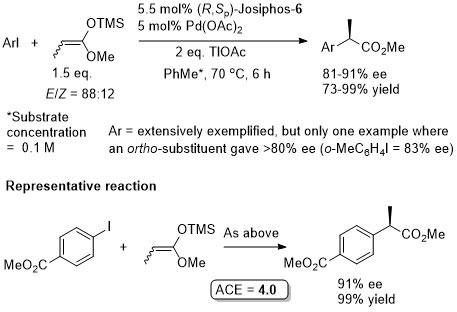
Pd – Cross-coupling Orgmet09-28-152
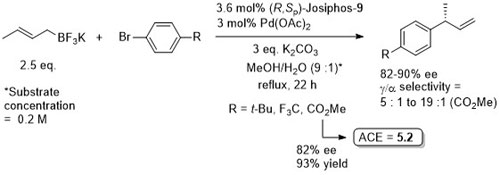
Pd – Cross-coupling/C-H activation JACS12-134-7305
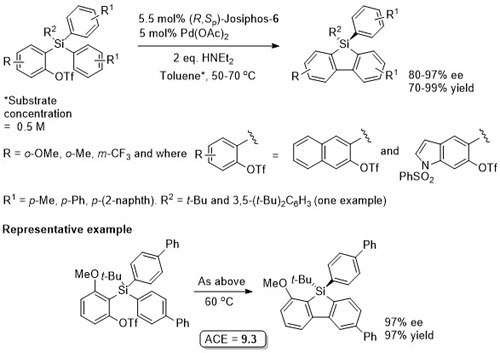
Pd – Carbonyl alpha-arylation Orgmet11-30-6323
Lower ee values with other R groups (and with OR = H), and also where one or both rings of the product spirocycle >5 membered.
Pd – Heck JOMC06-691-2159

The absolute configuration of the product was not established. Relative configuration confirmed by X-ray crystallography.
Pd – Hydrogenation CC11-47-5052
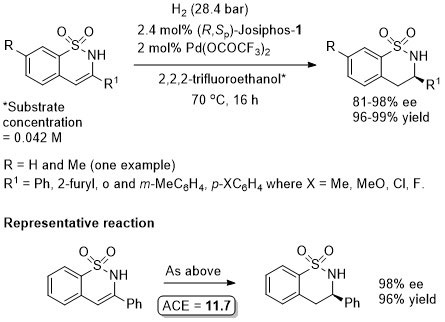
Hydrogenation proceeds following in situ tautomerisation to the corresponding cyclic N-sulfonylimine. Where R1 = alkyl <80% ee obtained, but with these substrates >90% ee was achieved with a Walphos ligand.
Pd – Reductive amination CC17-53-1704
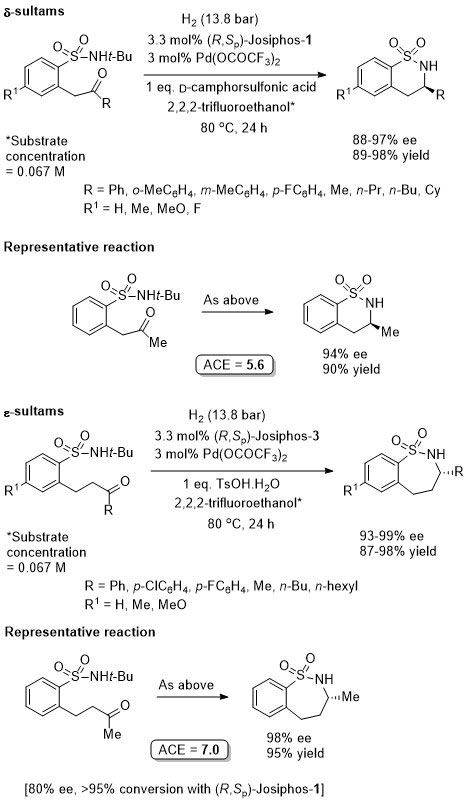
Pd – Methoxycarbonylation Helv06-89-1610
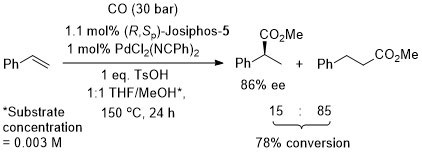
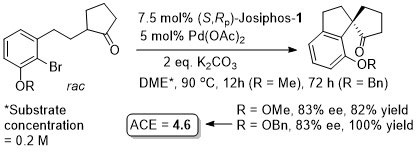
Pd – Desymmetrisation JACS04-126-10248
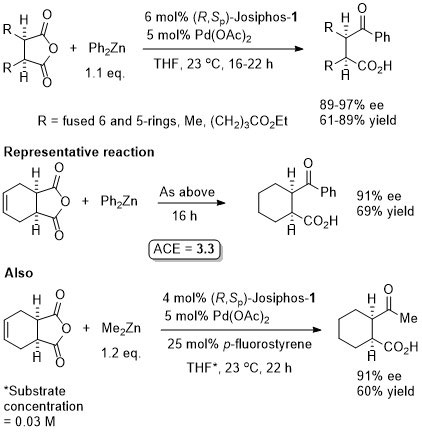
Ruthenium
Ru – Hydrogenation Chirality00-12-514
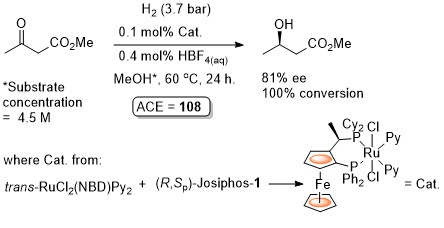
The catalyst formed in this way (and presumably used in the hydrogenation reaction) contained 10% of an unidentified compound. The corresponding catalyst derived from (R)-BINAP gave 99.9% ee (R selectivity) and 100% conversion for the same reaction under the same conditions.
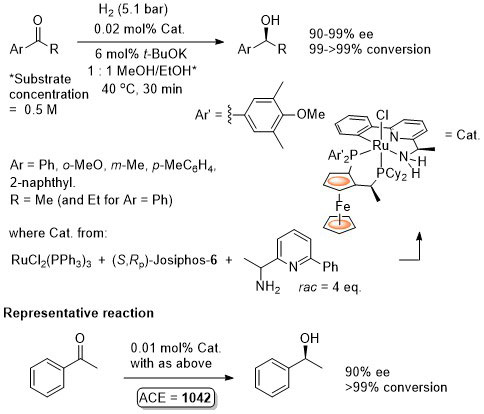
Predominently one enantiomer (R) of the pyridylamine is incorporated into the catalyst formed as >93% major isomer. Use of the (R)-amine (60% ee – 2.5 eq.) gives a single isomer of the catalyst. Also stated that there is no difference in the ‘activity’ of the complexes prepared by these two methods.
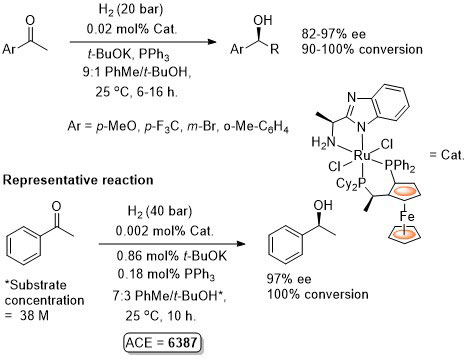
Catalyst prepared as described in CEJ08-14-9148
Ru – Transfer Hydrogenation Orgmet05-24-1660
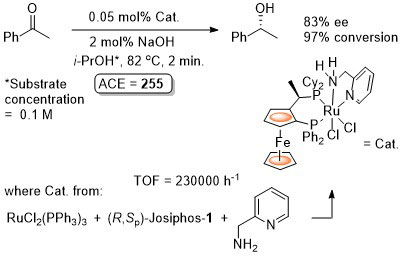
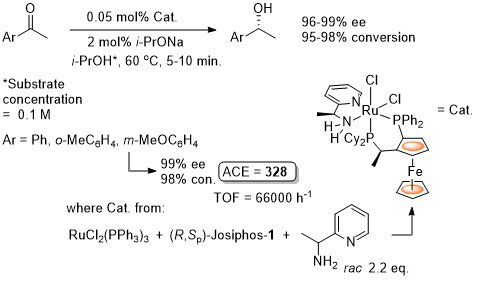
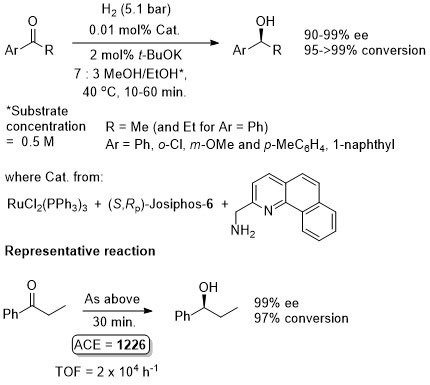
It is noted that “much the same” enantioselectivity was observed with an in situ generated catalyst. Similar enantioselectivities observed where the catalyst used (following isolation) was derived from: i) replacement of Me attached to the aminopyridine stereogenic centre by Ph or t-Bu and/or ii) use of Josiphos-6 instead of Josiphos-1.
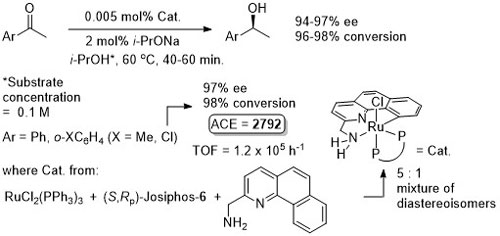
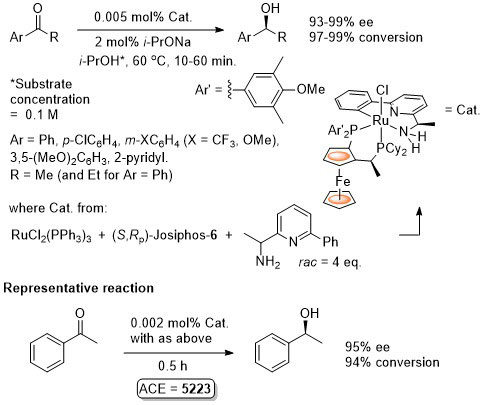
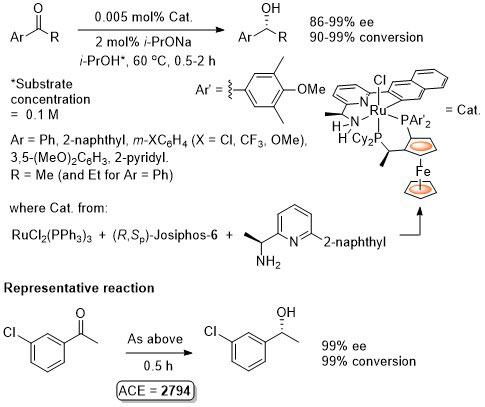
Catalyst also generated in situ to give similar enantioselectivity and activity (generally with a catalyst loading of 0.01 mol%).
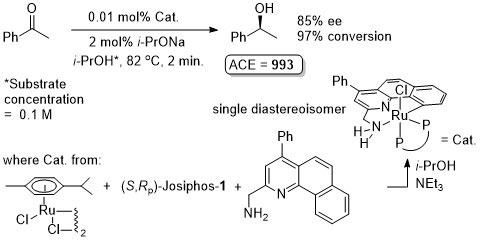
Osmium
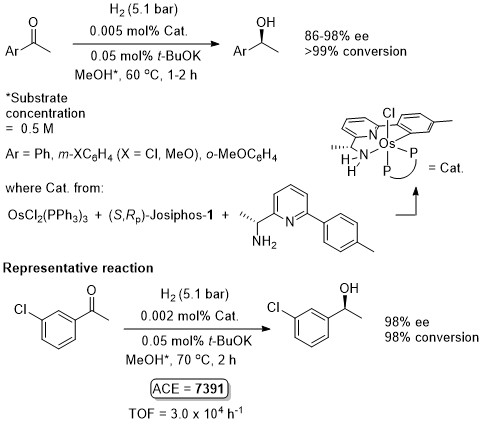
Os – Hydrogenation Angew08-47-4362
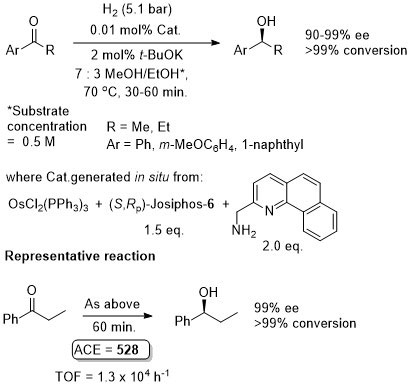
Os – Transfer Hydrogenation CEJ08-14-2557
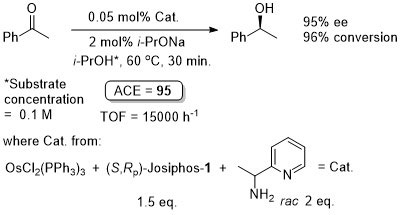
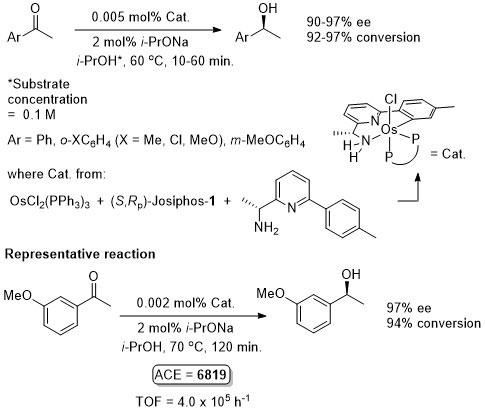
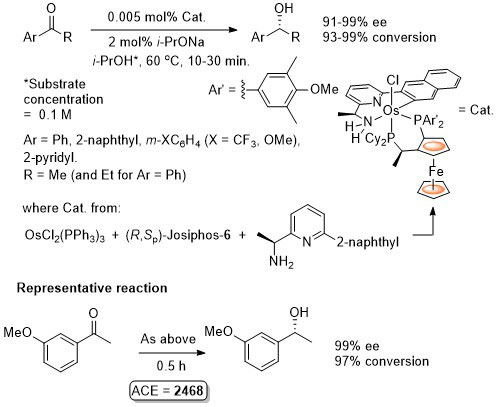
Catalyst also generated in situ to give similar enantioselectivity and activity (generally with a catalyst loading of 0.01 mol%).
Rhodium
Rh – Hydrogenation JACS94-116-4062
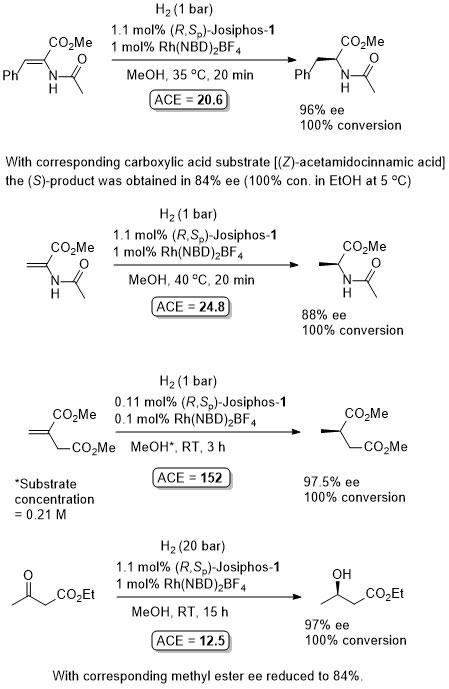
ASC08-350-898. The hydrogenation of methyl Z-α-acetamidocinnamate (MAC i.e. the first substrate above) results in a decrease in ee with an increase in hydrogen pressure. Orgmet07-26-3530. Hydrogenation of MAC (94% ee, 100% conversion) and dimethylitaconate (DMI – 99.5% ee, 100% conversion) under similar conditions (0.5 mol% Rh(NBD)2BF4, 0.525 mol% (R,Sp)-Josiphos-1, MeOH (substrate concentration = 0.25 M), H2 (1 bar) 25 oC, 1h). Product configurations are opposite to those given above. With (R,Sp)-Josiphos-5 DMI gave 92% ee, 96% conversion under these conditions. ASC06-348-1605. (R,Sp)-Josiphos-1/Rh(cod)2BF4 supported on alumina (CATAXA) with a phosphotungstic acid linker has been used for the hydrogention (160 bar) of DMI using continuous flow supercritical carbon dioxide as the reaction medium (83% ee, 24% conversion 55 oC). The absolute configuration of the product given in the graphical abstract (the only place where stated) is S, i.e. not R as given above. EJOC05-1909. Cat. From 1 mol% (R,Sp)-Josiphos-1 and 1 mol% Rh(cod)2BF4. Using this DMI (MeOH, substrate concentration = 0.21 M, H2 = 1.2 bar, 4 h, 70 oC) gave (S)-product in 99% ee, 100% conversion. Product configuration opposite to above scheme. Using MAC (MeOH, 0.21 M, H2 = 2.4 bar, 4 h, 110 oC) gave (S)-product in 74% ee, 100% conversion. Orgmet02-21-1766. Cat. From 0.5 mol% (R,Sp)-Josiphos-1 and 0.5 mol% Rh(NBD)2BF4. Using this DMI (MeOH, substrate concentration = 0.25 M, H2 = 1 bar, 22 h, 18 oC) gave (S)-product in 98.5% ee, 100% conversion. Product configuration opposite to above scheme. Using MAC (MeOH, 0.25 M, H2 = 1 bar, 66 h, 18 oC) gave (S)-product in 84.4% ee, 100% conversion. Angew07-46-5971. Propylene carbonate has been used as the solvent for the hydrogenation of dimethyl itaconate in 99% ee (1 mol% [Rh(cod)2]BF4/Josiphos-1, H2 1 bar, 25 oC), and under the same conditions MeOH and THF resulted in 88 and 92% ee respectively.
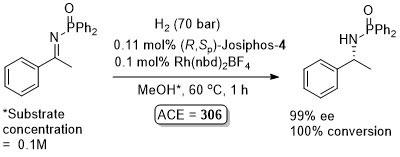
With 1 mol% catalyst loading also applied to corresponding p-Me (97% ee, 100% conversion, 21 h) and p-CF3 (93% ee, 98% conversion, 18 h) substrates. The reaction is sensitive to the identity of the p-substituent (e.g. 30% ee with p-Cl).
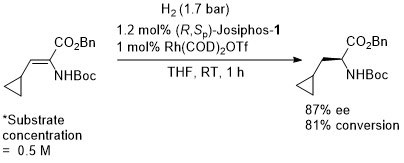
Result from a screening study identifying (R,Sp)-BoPhoz as a better ligand (98.6% ee and 100% conversion using the same conditions. (S,S)-Ethyl DuPhos also successful (99.3% ee and 100% conversion).
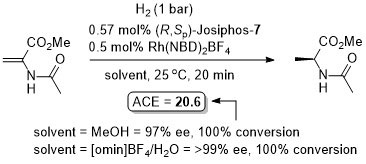
Absolute configuration not stated so assigned by comparison to the result in JACS94-116-4062. With the [omin]BF4/H2O mixture (substrate 0.25 M wrt H2O and approx. 1:1 ratio by volume of ionic liquid/water) the catalyst (following decantation of aqueous phase) was reused six times with a 70% conversion from the final run. Essentially identical results reported with this substrate in ASC07-349-1803 (TBME 99% ee, tolene 98% ee, i-PrOH 97% ee – and as a 1:1 mixture with 1:1 [bmim]BF4).
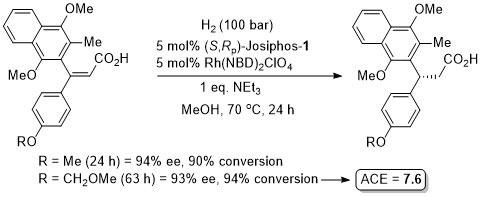
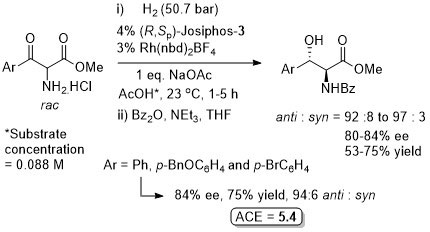
A dynamic kinetic resolution. Other Ar substituents gave <80% ee.
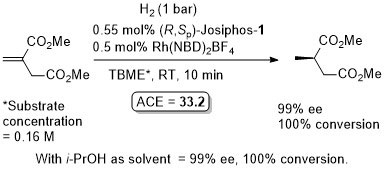
Absolute configuration not stated so assigned by comparison to the result in JACS94-116-4062. Use of (R,Sp)-Josiphos-7 in i-PrOH also gave 99% ee, 100% conversion. Essentially identical ees were obtained when both ligands (and both solvents for 1) were used with 1:1 [bmim]BF4.
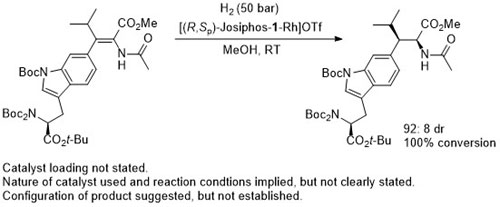
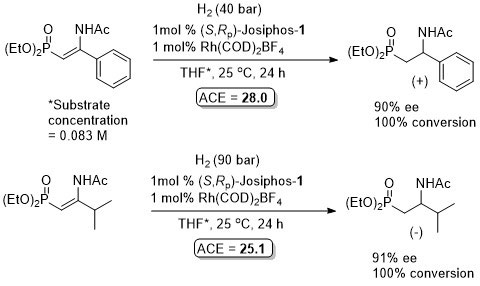
Absolute configuration not give. It is stated that “a comparison with the specific rotation of known chiral β-substituted β-amino-ethan-1-ylphosphonates indicates that a +ve sign correlates with an (S)-configuration.” Of note that the use of (R,Sp)-Josiphos-1 with the dimethyl phosphonate analogue of the substrate for the first of these examples is also reported to give a product with a positive rotation. See TA09-20-1437.
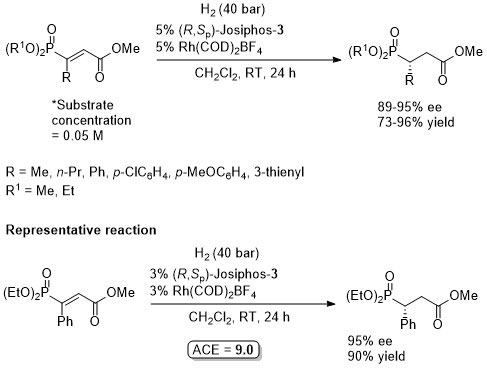
Absolute configuration assigned by comparison to data in CEJ09-15-10983. Mandyphos-2 has also been applied to this reaction.
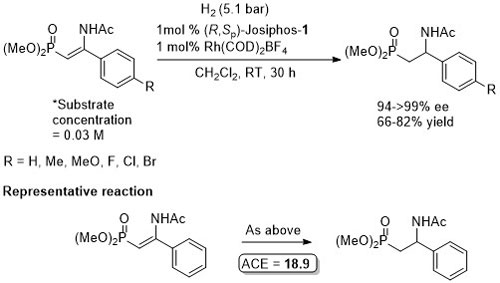
Absolute configuration assigned tentatively as S. Some of these results also reported in Orgmet09-28-888.
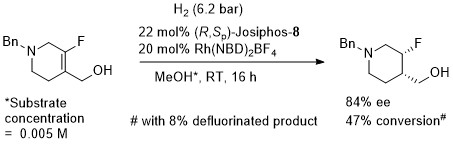
Result from a ligand scoping study from which the best results were obtained with a Walphos ligand.
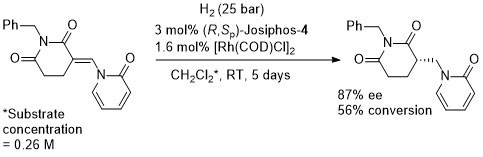
Opposite (R) configured product obtained with (R,SP)-Josiphos-1 (76% ee). Higher ee values obtained with (R,R)-EtDuPHOS [(S)-product)] and a Taniaphos type ligand [(R)-product with further reduction to a 2-piperidinone, 98% ee].
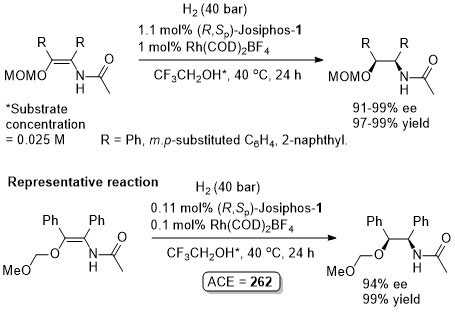
Rh – Imine hydrogenation OL13-15-484
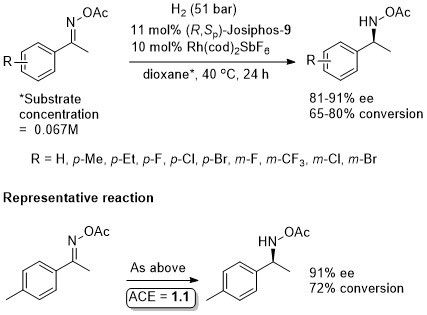
Also several examples with ee <80%. Low values (~40% ee) with ortho-substituents. From the method in the SI the catalyst loading is 1 mol% and not 10 mol% as above. As the 10 mol% loading is used throughout the paper this is used above, but it is possible the authors have done themselves a disservice.
Rh – Isomerisation Helv01-84-230
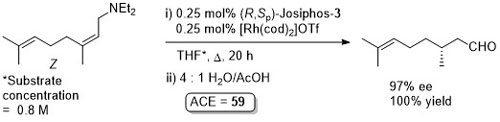
Under the same conditions the corresponding E configured substrate gives the S product in 92% ee and 99% yield.
Rh – Hydroboration JACS94-116-4062
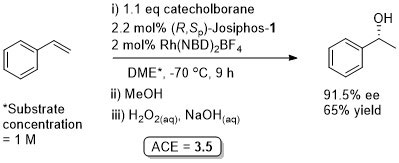
Less than 1% of regioisomeric 2-phenylethanol formed. Relatively low reactivity in comparison to bis(diphenylphosphino) ligands due to the higher basicity of Josiphos-1.
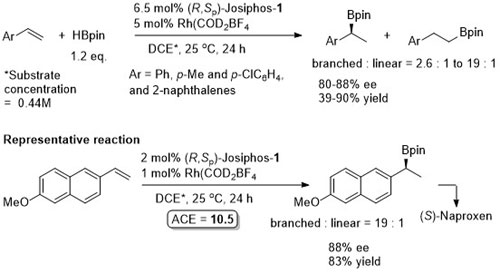
Use of HBpin gives opposite enantioselection to that obtained with catacol borane (HBcat). HBPin is more stable and easier to use than HBcat, but obtaining the branched isomer is more challenging with the latter reagent.
Rh – Asymmetric ring-opening JACS00-122-5650
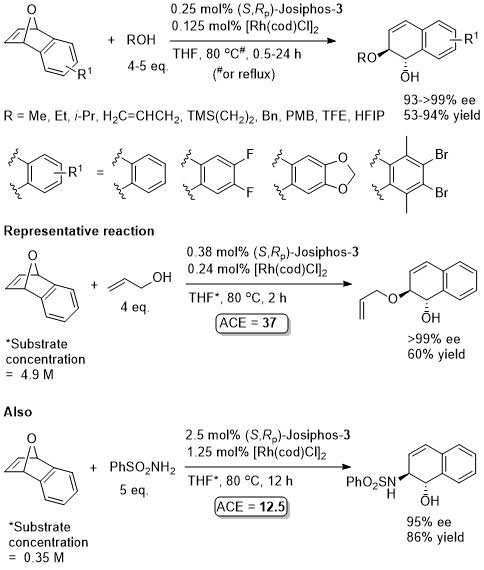
The reactions with MeOH, EtOH and i-PrOH were with 10 eq. of the alcohol and 1 mol% of the rhodium catalyst, but there is nothing to indicate they could not be carried out with the lower catalyst loading indicated. The actual catalyst loadings used (from SI) are variable (see representative example). There is significant confusion in both the paper and the SI of the correlation between the catalyst and product configurations. There are assigned using later papers from this group. The nitrogen example is the only example where the ee is >80% (others 45-74% ee). For MeOH an ee of 88% is reported in PNAS04-101-5455 with use of Josiphos-1 (compared to 96% with Josiphos-3), this paper also describes a mechanistic model. For MeOH/Josiphos-1 an ee of 92.4% (36% yield) was obtained by another group – see JOMC05-690-1166 (0.5 mol% [Rh(cod)Cl]2, 1 mol% ligand, 7 eq. MeOH, THF (substrate concentration 2.3 M), reaction heated [presumably reflux?] 5 h). The results with alcohols as nucleophiles are repeated in JOMC01-624-259. The only significant difference is that the yield with allyl alcohol as nucleophile is 92% (>99% ee) for which ACE = 55.9.
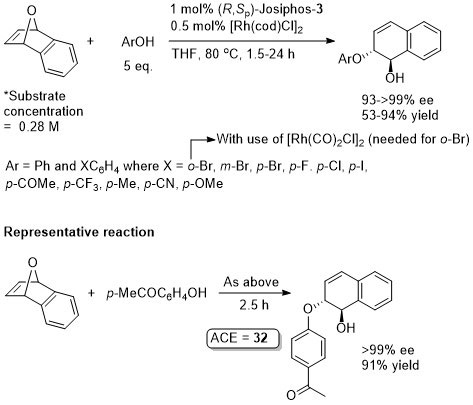
More acidic phenols observed to add faster. The results are essentially the same as repeated in JOMC01-624-259.

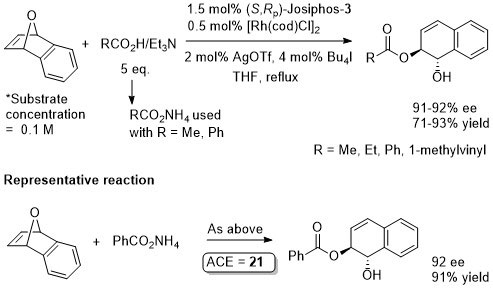
Just (S,Rp)-Josiphos-3 and [Rh(cod)Cl]2 used for the calculation of ACE. Following the combination of these the catalyst was generated in situ by chloride to iodide ligand exchanges using AgOTf then Bu4NI. Reactions quantities and results given are taken from the SI which differ a little from the results presented in the paper (e.g. concentration = 1.0 M from SI but 0.2 M in paper). No preactivation by deprotonation of the malonates is required. Also given, with oxabenzonorbornadiene as the substate, are results with p-methoxybenzylamine (81% ee, 71% yield) and dibenzylamine (88% ee, 91% yield), although the exact conditions used is not entirely clear.
Just (S,Rp)-Josiphos-3 and [Rh(cod)Cl]2 used for the calculation of ACE. Following the combination of these the catalyst was generated in situ by chloride to iodide ligand exchanges using AgOTf then Bu4NI.
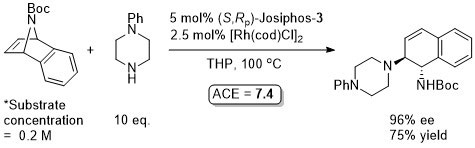
Just one example with Josiphos-3 which is incorrectly given in the paper as the (R,Sp)-enantiomer (i.e. correct in the abstact above). Reaction further exemplified (also with pyrrolidine, piperidine, morpholine and dibenzylamine as the nucleophile) using as ligand (S,S,Rp,Rp)-2,2′-bis(α-N,N-dimethylaminoethyl)-1,1′-bis(diphenylphosphino)ferrocene (a type of Ferriphos ligand that is not commercially available). In the paper this is incorrectly drawn as the (R,R,Sp,Sp)-enantiomer. For more details see JACS06-128-6837.
Rh – Asymmetric ring-opening (+isomerisation and oxidation) Angew11-50-7346
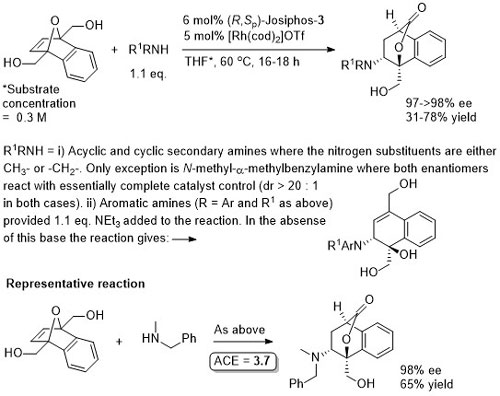
Rh – Epoxide isomerisation Angew17-56-6307
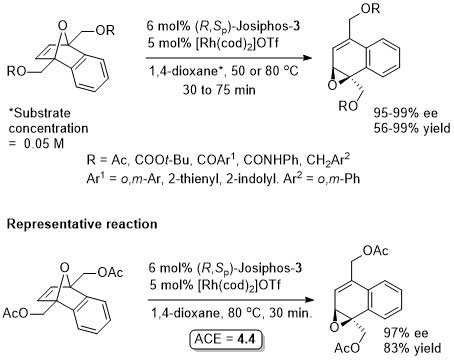
Rh – Asymmetric ring-opening – diazabicycle desymmetrisation Angew08-47-2085
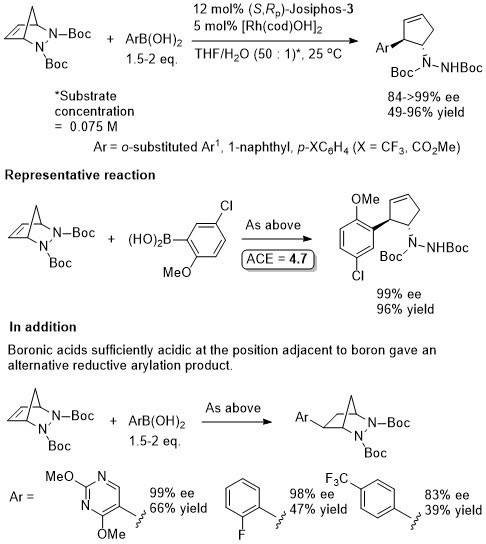
Rh – Hydroamination of allenes CS16-7-3313
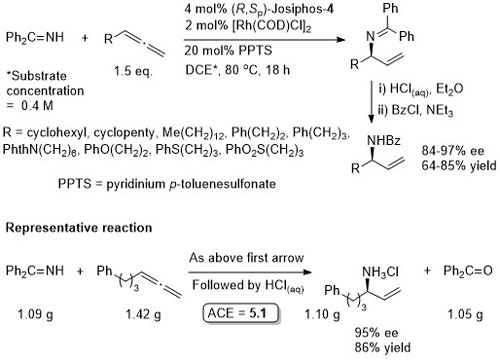
Rh – Conjugate addition EJOC02-3552
Conditions stated are the optimised conditions with which the reaction was exemplified with (R)-BINAP (16 examples). For this example (S,Rp)-Josiphos-1 gave the same ee (and configuration) and the implication is these conditions were also used. Water is crucial for the reaction as in its absence the reaction is very slow and the ee significantly reduced.
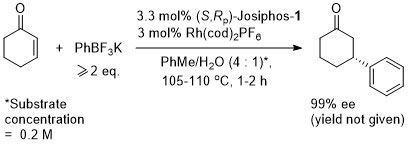
Rh – Hydroalkynylation OL13-15-5956
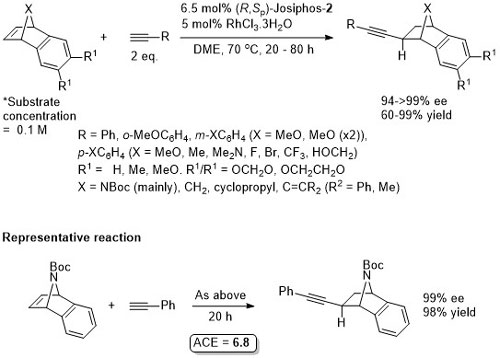
Iridium
Ir – Hydrogenation
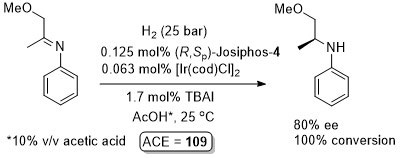
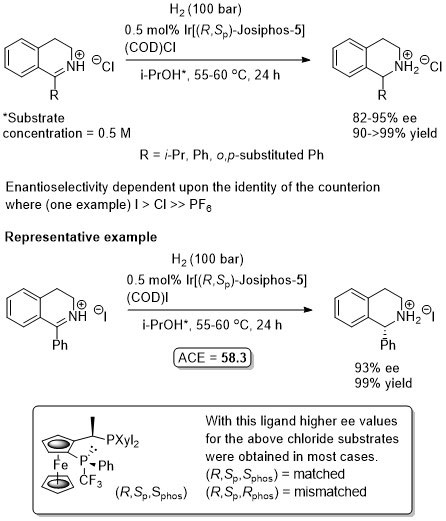
Substrate/catalyst (s/c) ratio given as 800, thus assumed this was generated from a 1:1 ligand:metal ratio (not explicitly stated in paper). A description of the extension of this reaction as the key process in the production of > 10000 t/y of (S)-metolachlor is given in Chimia99-53-275. Optimised conditions = H2 (80 bar), 50 oC, s/c > 1000000, reaction time = 4 h, 79% ee, 100% conversion. Other exact details of the process are not given, but assuming the catalyst is generated from the same ratio of ligand/metal components in the scheme, and for s/c = 1000000, ACE = 133970. Stated that it proved possible to further decrease the catalyst loading to give a s/c ratio of 2000000.
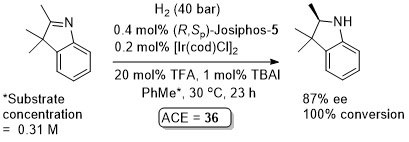
Absolute configuration of product not stated so assigned by comparison to the use of the same (R,Sp)-ligand for the synthesis of (S)-Metolachor (Chimia99-53-275). Substrate/catalyst ratio given as 250 thus assumed this was generated from a 1:1 ratio ligand:metal (not explicitly stated in paper).
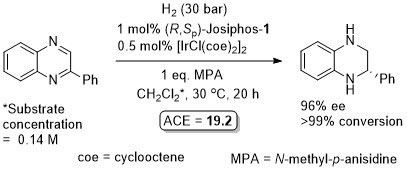
Copper
Cu – 1,4-Grignard Conjugate addition
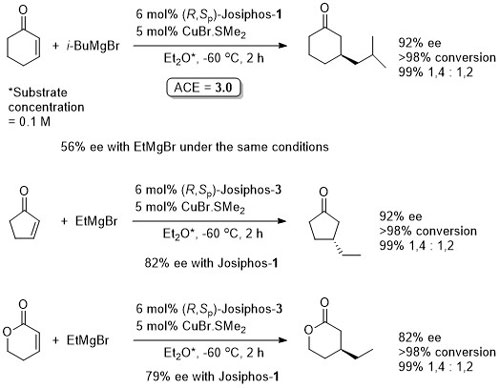
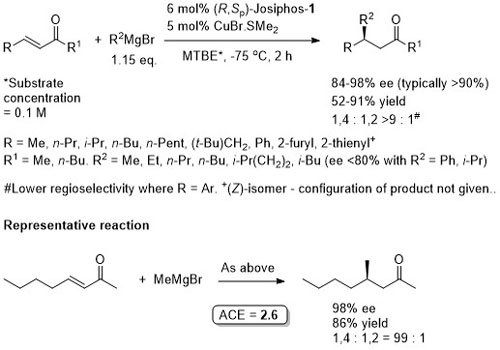
Methodology applied to the asymmetric total synthesis of metacycloprodigiosin and prodigiosin R1. See JACS09-131-14579.
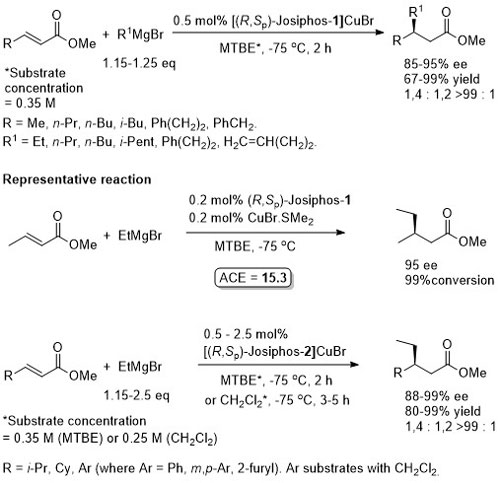
No difference between in situ formed and preformed catalyst. Very slow reaction with MeMgBr. Josiphos-2 better for bulky/aryl beta-substituents.
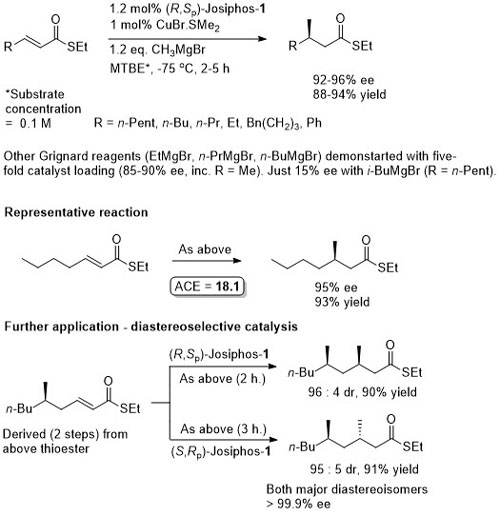
Thioesters more reactive towards conjugate addition than corresponding oxoesters (closer to enones). Basis of an iterative methodology demonstrated with the synthesis of (-)-lardolure. Also applied to the synthesis of mycocerosic acid, phthioceranic acid, a putative wasp pheromone, a simplified analogues of caspofungin, mycolipenic acid, glycolipid antigens and tuberculostearic acid. See also the synthesis of β-D-mannosyl phosphomycoketides and neopeltolide.
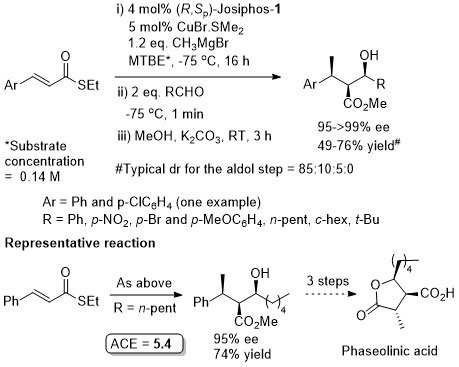
Conversion of the crude aldol product to the oxygen ester gave greater stability for diastereoisomer purification (column chromatography and in some cases crystallisation). Use of an alkyl β–substituent in the unsaturated thioester substrate (CH2OTBDPS) resulted in poor aldol diastereoselectivity (~1:1).
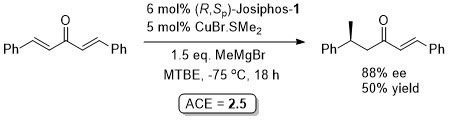
A higher ee value but a lower yield was obtained from the use of a (S,R,R)-phosphoramidite ligand with Me2Zn [95% ee (S), 12% yield] or AlMe3 [96% ee (R), 16% yield].
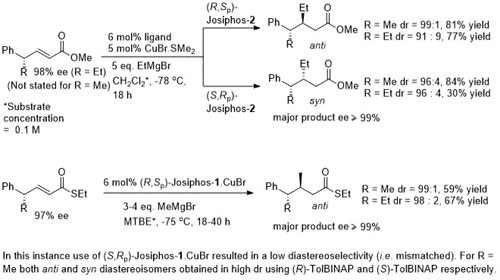
Josiphos-1 was also applied to reaction of the corresponding methyl ketone (R = Me). (R,Sp)- Josiphos-1 resulted in the anti isomer in high dr (92 : 8 with EtMgBr and 98:2 with MeMgBr). In contrast (S,Rp)-Josiphos-1 resulted in poor syn selectivity.
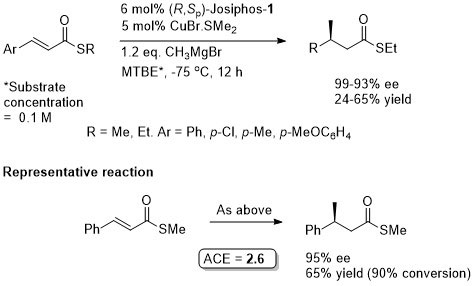
Substrates containing an aryl substituent at the beta position are generally less reactive towards conjugate addition with Grignard reagents. This paper goes on to report that higher activities and high enantiomeric excesses were observed with Tol-BINAP as ligand.
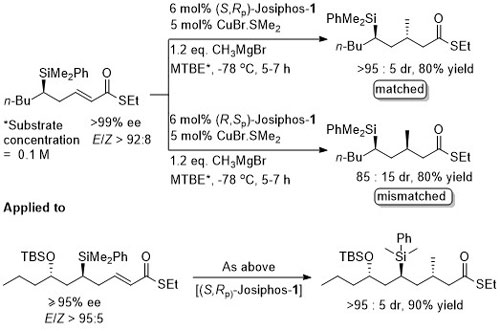
The SiMe2Ph may be replaced (with retention of configuration) by a hydroxyl group using a Tamao-Fleming oxidation.
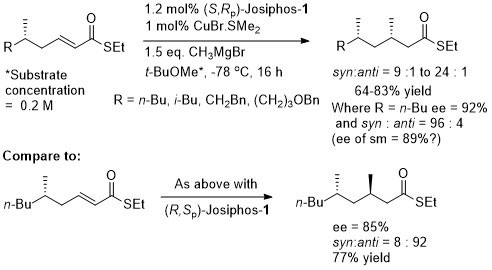
Suggested that the low diastereoselectivity a result of TiCl4 mediated epimerisation.
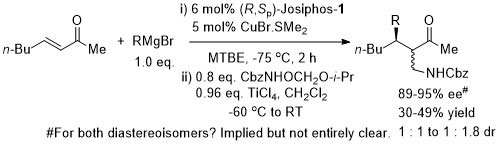
Low yield and low ee obtained with MeMgBr. Traping of the magnesium enolate formed in the representative reaction with PhCHO resulted in the trans product as a 1.15 : 1 ratio of diastereoisomers (i.e. epimeric at -CHPhOH stereogenic centre).
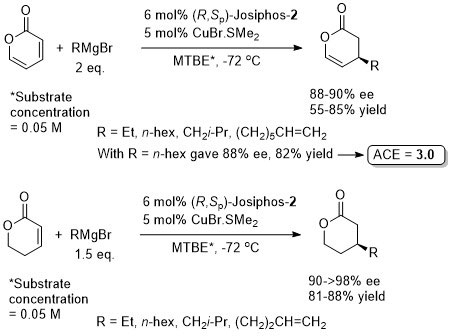
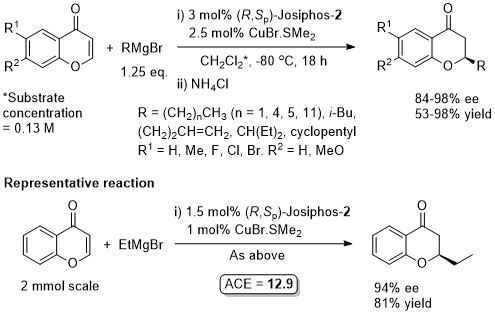
The 2H-pyran-2-one substrate is less reactive than acyclic α,β-unsaturated esters towards conjugate addition (due to increased electron delocalisation). The higher reactivity of the 5,6-dihydro-2H-pyran-2-one substrate enabled a reduction in the equivalents of Grignard reagent employed from 2 to 1.5. With the more reactive substrate MeMgBr gave the addition product but in low ee (50% ee, 68% yield).
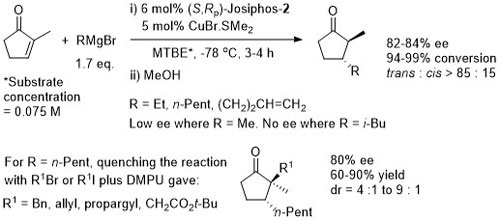
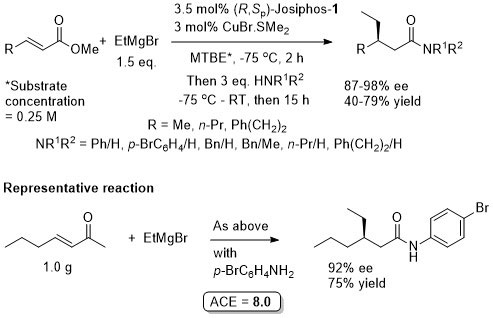
Also one example with n-HexMgBr. Less reactive MeMgBr was used with (S)-Tol-Binap and CuI as reported in ASC08-350-673. The configuration of Josiphos given in this paper is (S,Rp) which must be incorrect for the configuration of the products reported. Also the loading of Josiphos and CuBr-SMe2 given in the discussion is 7.5 mol% and 5 mol% respectively. The loadings given in the experimental are different, and are used above (to give the benefit of the doubt).
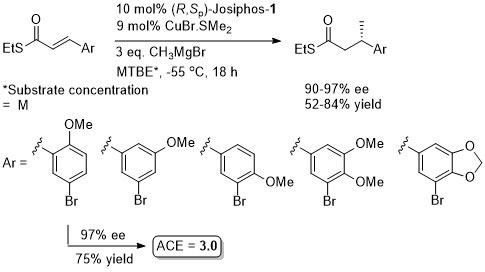
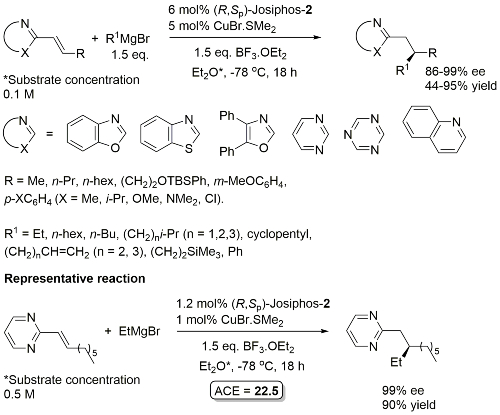

TMSOTf with unreactive, unhindered amides. BF3.OEt2 with reactive amides (hindered and unhindered). Low conversions/racemic products with primary or secondry amides. Traces of THF detrimental to conversion/enantioselectivity. It is not stated how the absolute configuration of the cyclic amide product was determined. By analogy to the reaction of cyclohexenone and 5,6-dihydro-2H-pyran-2-one this product might be expected to have the opposite configuration (i.e. S) to that given in the scheme above. See PNAS04-101-5837.
Cu – Grignard 1,6-Conjugate addition
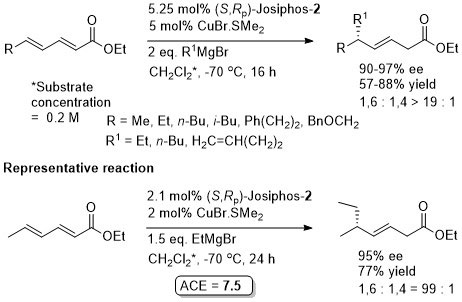
Little (<10%) reaction with MeMgBr.
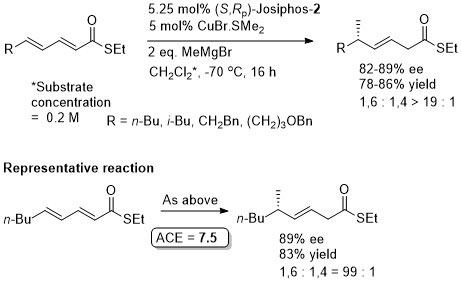
Cu – Grignard addition to cyclopropenes
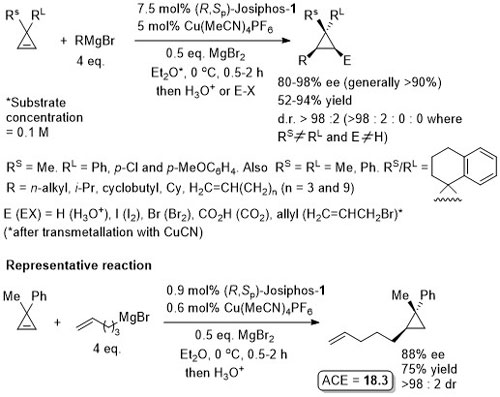
Cu – Organoaluminium/Zinc Conjugate addition
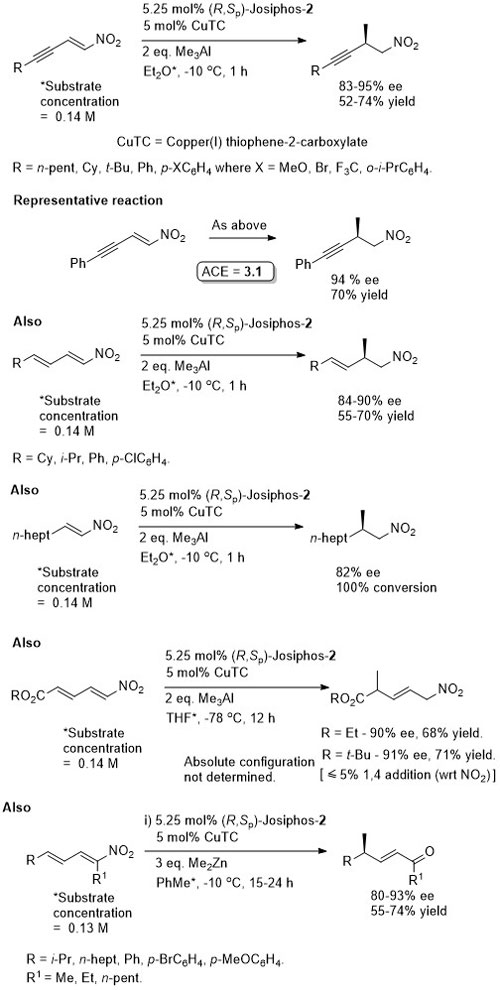
Cu – Enolate Conjugate addition (Michael Reaction)
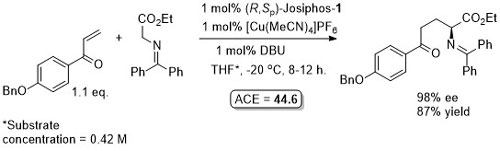
This reaction was exemplified and extended using phosferrox ligand PN-L1a which also resulted in high enantioselectivity.
Cu – Grignard addition to ketones
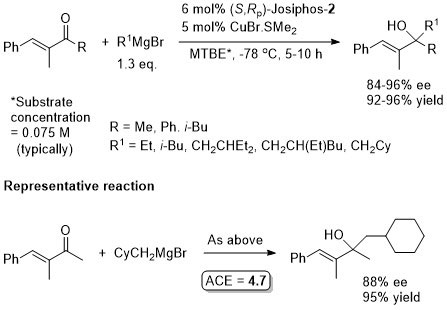
No reaction with MeMgBr and a racemic product obtained with PhMgBr. Absolute configuration not stated.
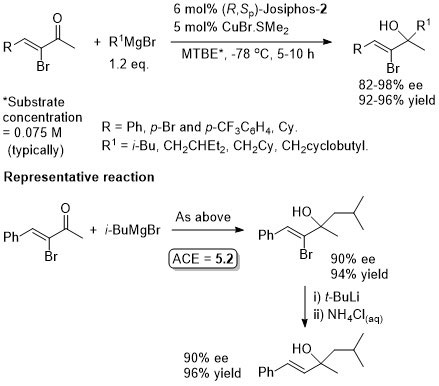
Absolute configurations are implied in the manuscript schemes but it is not stated how determined. However the supporting information states that the absolute configurations were not determined, thus not given here. A marked positive non-linear effect (asymmetric amplification) has been reported for this reaction due to the selective precipitation of the meso (R,SP,S,RP) dimer under the reaction conditions. See: CC13-49-5450.
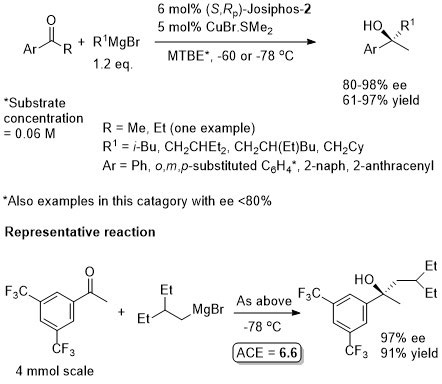
Absolute configuration assigned by comparison of the sign of the optical rotation of the products with that of (R)-2- phenylbutan-2-ol, a compound not synthesised using this methodology.
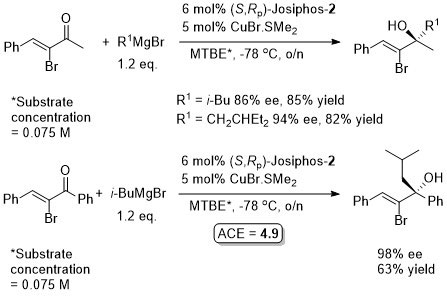
Absolute configurations determined after Br to H conversion and oxidation to corresponding α-hydroxyacid. Revealed that the methyl ketone and the phenyl ketone substrates have opposite facial selectivity.

Absolute configuration not determined. Assigned as R by comparison to substrates studied previously.
Cu – Grignard addition to acylsilanes
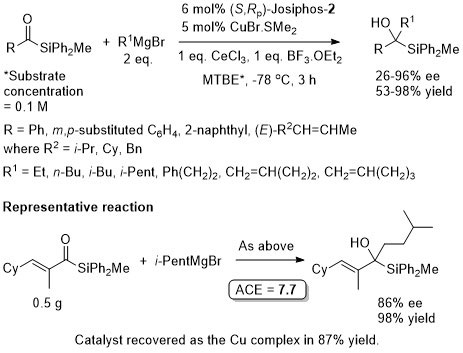
Absolute configuration not stated. Low conversion and racemic product obtained with MeMgBr. For absolute configuration determination see Angew16-55-6057.
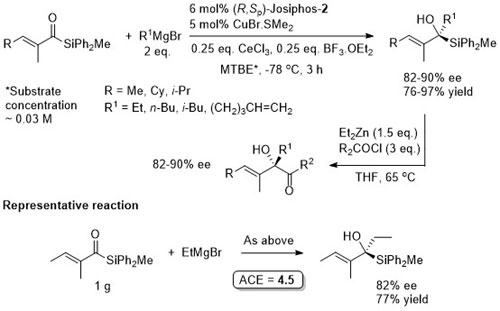
Absolute configuration established. Reaction of the product of Grignard addition with diethylzinc resulted in a zinc-Brook rearrangement followed by addition of the allylzinc intermediate an acid chloride (as electrophile). The overall sequence gave complete chirality transfer.
Cu – Addition of Grignard reagents to ketimes
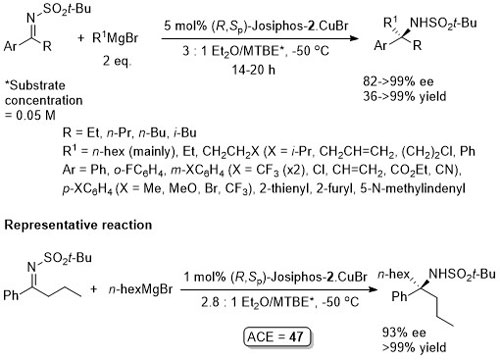
Addition to corresponding dialkyl ketimines was unsuccessful. Use of methylmagnesium bromide gave a racemic addition product and use of substrates with R = Me resulted in ee values <80%.
Cu – Grignard allylic substitution
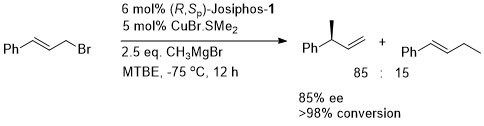
Higher ee values obtained with a Taniaphos ligand. Absolute configuration assigned by comparison to results reported in JACS06-128-15572.
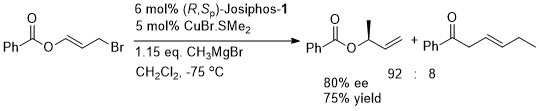
Higher ee values obtained with a Taniaphos ligand.
Cu – SN2′ Grignard addition to 1,3-cyclohexadiene monoepoxide Synlett07-435
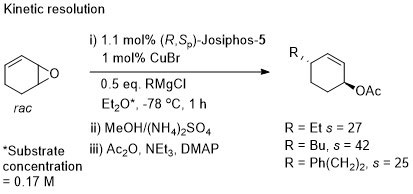
The product was much easier to isolate as its acetate. The use of alkylmagnesium bromides resulted in competitive bromide ion attack on the substrate.
Cu – Hydroboration

Cu – Beta (1,4) borylation
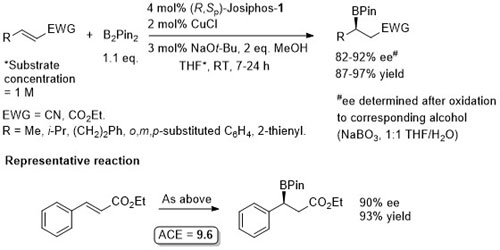
Similar ee values obtained with Mandyphos-1. A preliminary result with Josiphos published in OL06-8-4887.
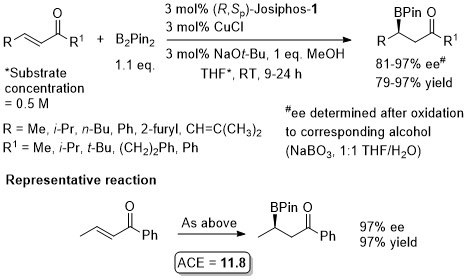
Mandyphos-1 also applied to this reaction.
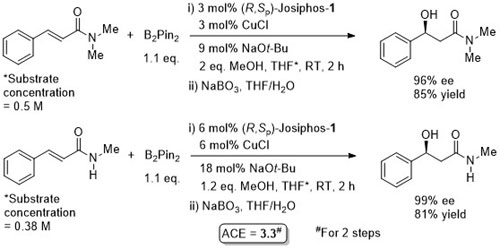
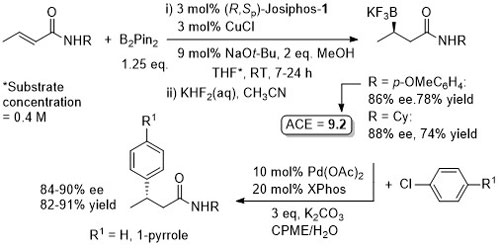
Inversion of stereochemistry on transmetallation to palladium a unique feature of the reactivity of beta-trifluoroboratoamides due to participation of the carbonyl oxygen.
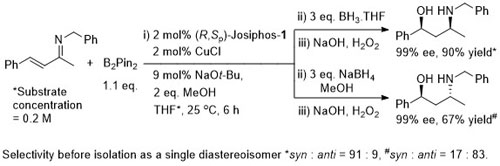
The syn N-n-butyl derivative was generated in the same way in 80% ee. The syn N-phenyl derivative was similarly generated in 92% ee, 69% yield using a phosphoramidite ligand.
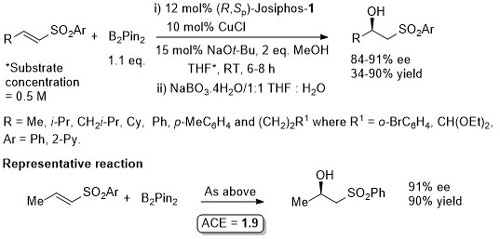
Similar ee values and the same absolute configuration obtained with (R,Rp)-Taniaphos-1. MeOH added to a solution containing all of the other species – this resulted in an increase in reactivity.
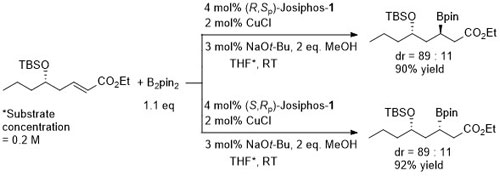
Complete catalyst control with imperfect asymmetric induction.
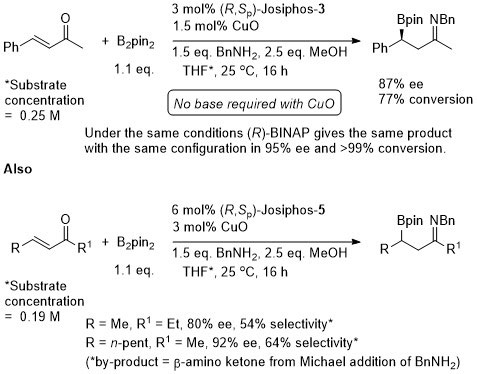
Absolute configuration not stated for the products resulting from the latter reaction.
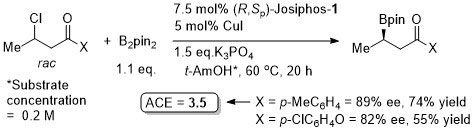
In situ generation of corresponding alpha,beta-unsaturated carbonyl compound. For enones higher ee values with a Taniaphos ligand.
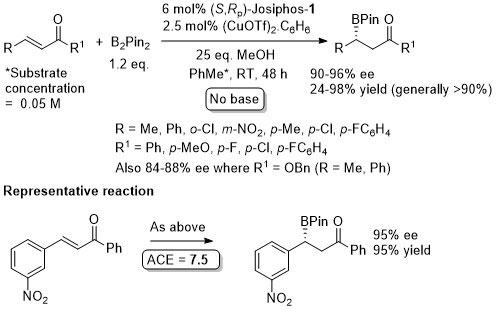
For R = Ph ee = 48% with Josiphos-1. Improved to 80% ee (product configuration = R) with (1R,2R)-N,N′-dimethyl-1,2-diphenylethane-1,2-diamine.
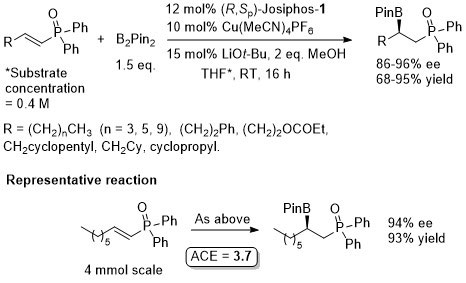
Lower enantioselectivity with strongly electron withdrawing substituents or, in general, ortho substituents.
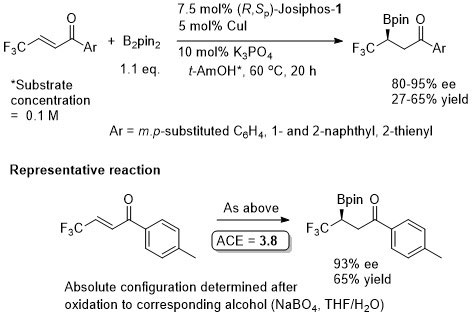
Starting materials given as >99% ee [from (R)-DM-BINAP β-borylation of in situ generated imine (from benzhydrylamine) of α,β-unsaturated aldehyde, followed by hydrolysis and Wittig reaction]. Low diastereoselectivity where R = Me or n-Pr. Note – the drawing of (S,Rp)-Josiphos in the manuscript is incorrect.
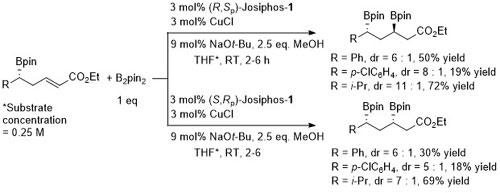
Cu – Beta (1,4) borylation and enolate trapping

Cu – Delta (1,6) borylation
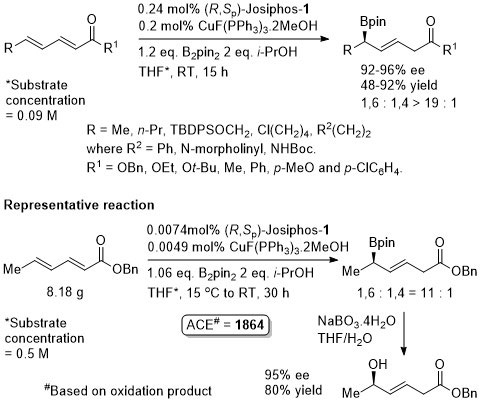
With R = Ph a complex mixture resulted. With R = Cy gave beta (1,4)-addition with the resulting alcohol following oxidation obtained in 87% ee.
Cu – Vinyl borylation
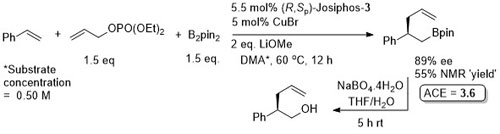
Use of (R,Sp)-Josiphos-1 at 40 oC resulted in an ee of 87% (73% NMR yield). The reaction was further exemplified using as ligand (R,R)-QuinoxP. Use of this for the reaction above under similar conditions, but at 40 oC, gave 88% ee (77% NMR yield), and use of CuCl (at 40 oC) gave 90% ee (78% NMR yield). It is not explicitly stated (although implied) that the absolute configuration of the product from (R,Sp)-Josiphos is as given in the scheme – this is from the use of (R,R)-QuinoxP. The structure give for (R,R)-QuinoxP in the paper is incorrect = (R,S)-QuinoxP.
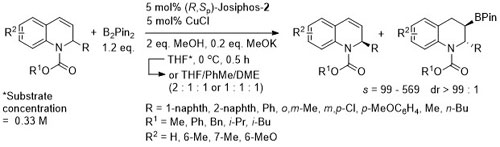
For one example (R = 1-naphth, R1 = Me, R2 = H) the catalyst loading was reduced to 0.3 mol% (at 10 oC, 12 h). Starting material = 44% yield, 99% ee. Product = 45% yield, 96% ee. The representation in the manuscript of the X-ray structure of the product of this reaction is of the wrong enantiomer (CIF file correct).
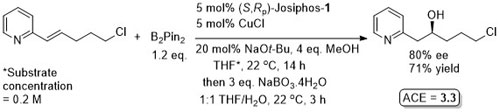
This example from a ligand screen. Higher ee (96% – same configuration of product implied) under these conditions with (R,R)-Ph-BPE, and with this ligand the reaction was exemplified with a range of 2-pyridyl and related N-heterocyclic substrates (position of nitrogen important for reactivity/enantioselectivity).
Cu – Beta (1,4) hydride addition (reduction)
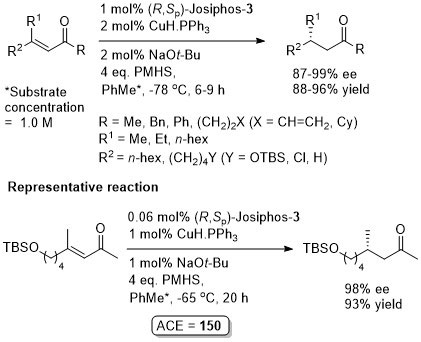
The representative procedure in the paper and the SI state CuH.PPh3 as the copper reagent used. In contrast the tables in the paper give this as CuCl. A Walphos ligand has also been applied successfully to a small number of examples.
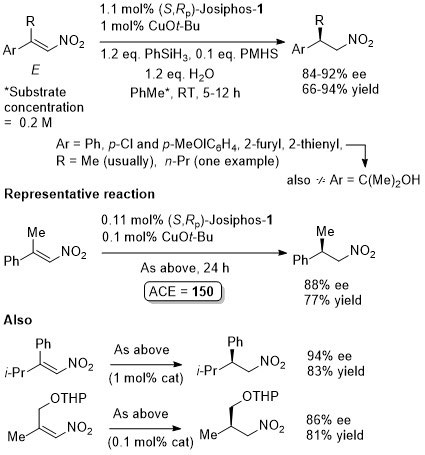
Reaction inhibited by halides – hence use of CuOt-Bu (although it is oxygen and moisture sensitive). Alternative methodology uses CuF2 (see OL04-6-4575).
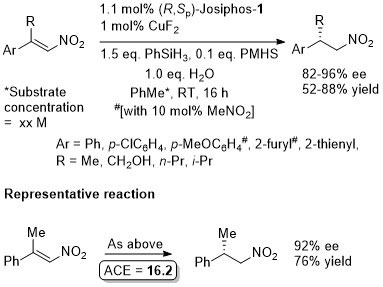
Suggested that fluoride sequestered by reaction with silanes (i.e. therefore no halide inhibition). Active catalyst a Cu(I) species generated in situ with substrates that are not electron rich. Nitromethane added to electron rich substrates to generate Cu(I) (possibly by deprotonation and oxidative coupling).
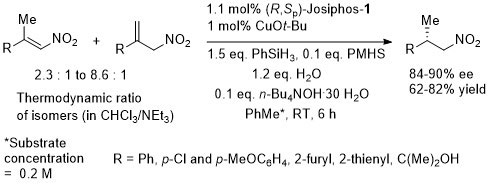
Trisubstituted nitoalkenes have a tendency to isomerise under basic conditions or on prolonged storage. This method avoids the need to separate these isomers. Ammonium base added after 2 h.
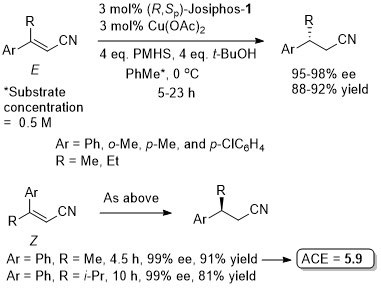
Synthesis07-2233 (essentially identical to the above paper)
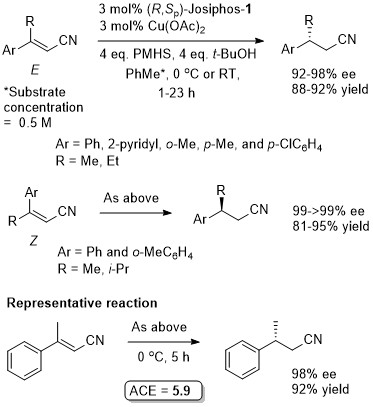
The absolute configuration of the product of the representative reaction is given as S which fits with other Cu-catalysed reductions with (R,Sp)-Josiphos-1. The structure of this product is incorrectly drawn as R in the supporting information.
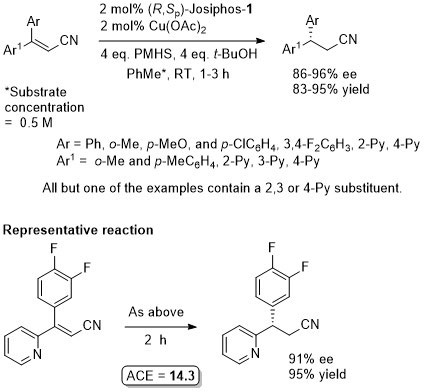
Under the same conditions the (E)-diastereoisomer of the substrate gave the S product in 86% ee, 91% yield.
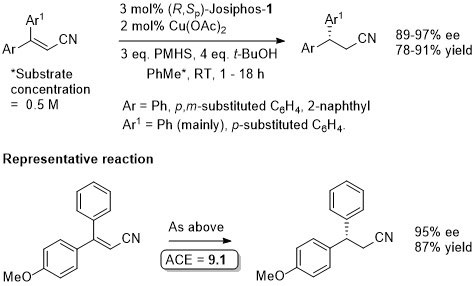
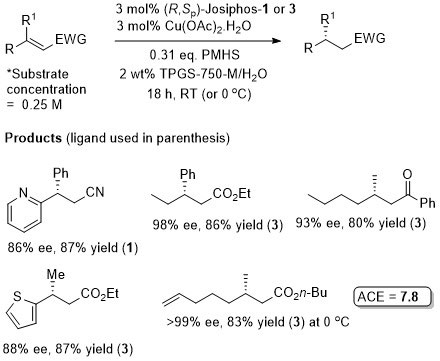
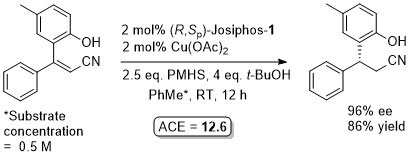
Micellar catalysis in water using surfactant TPGS-750-M with PMHS as hydride source.
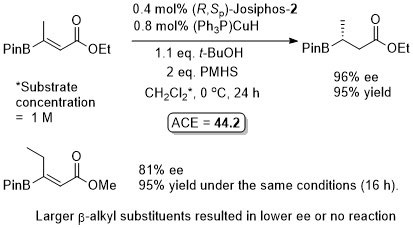
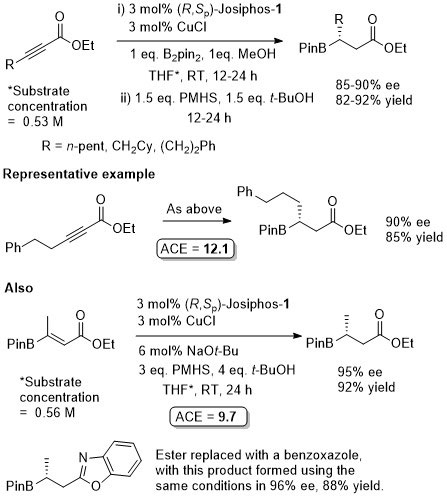
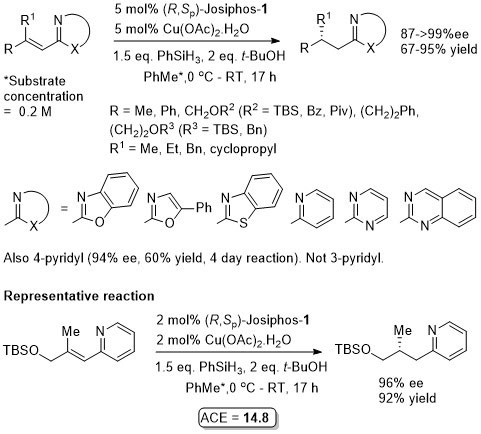
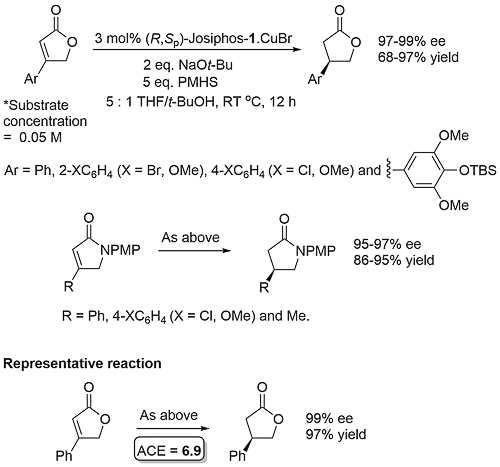
Josiphos-1.CuBr used as a preformed complex. The (Z)-acyclic precursors to the lactams PMP(Boc)NCH2ArC=CHCO2Et were reduced similarly with lower ee (83-85% ee – same sense of enantioselectivity).
Cu – Carbonyl reduction

Absolute configuration not given. Result from a ligand screening exercise. Best results with (R)-DTBM-SEGPHOS – starting with acetophenone (55 g scale) gave (R)-product using (R)-DTBM-SEGPHOS (0.001 mol%), CuCl (0.5 mol%), NaOt-Bu (0.5 mol%), PMHS (4 eq.), PhMe, -78 oC, 4 days. Yield = 73%, ee = 96%, ACE = 169.
Cu – 1,3-Enyne/aldehyde coupling
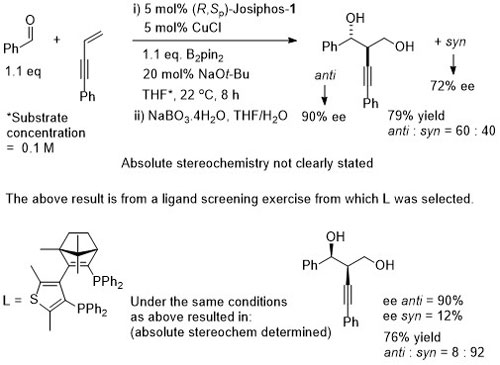
Cobalt
Silylacetylene addition to 1,1-disubstituted allenes
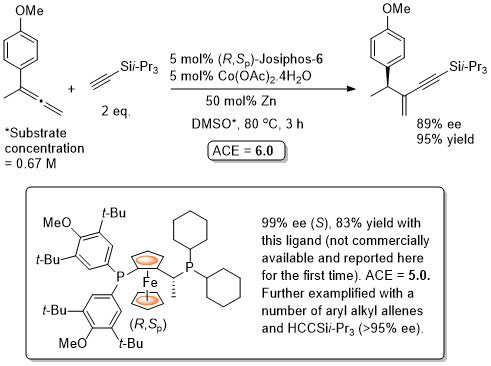
Nickel
Hydrogenation ChemCatChem09-1-237
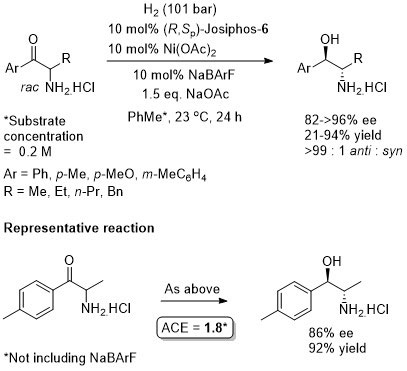
A dynamic kinetic resolution.
Allylic amination OL04-6-2661
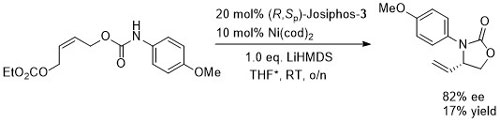
Ligand screening gave the highest ee with (R,Sp)-Josiphos-3 but a significantly higher yield (75% ee, 83% yield of (S)-product) with (R)-MeO-BIPHEP. No base was required for this reaction.
Organocatalysis