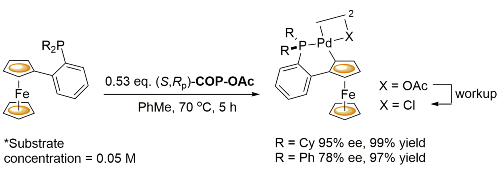
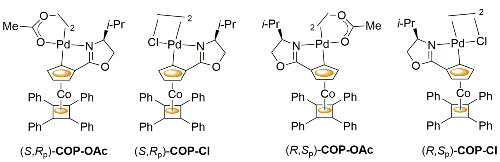
COP-OAc first reported by Richards and Stevens generated by diastereoselective palladation with Pd(OAc)2. COP-Cl and other COP derivatives generated by ligand exchange. See OS07-84-139 and OS07-84-148. Reviews on the application of COP catalysts available as ACR16-49-2220 and OS18-95-500. The following lists reactions for which the application of these palladacycle catalysts has resulted in >80% ee. ACE = Asymmetric Catalytic Efficiency. Literature covered until 2022 (24 entries). Previously, but no longer commercially available [e.g. (R)-COP-Cl, (R)-COP-OAc)]. Available within the group.
Pd – Allylic Imidate Rearrangement OL03-5-1809
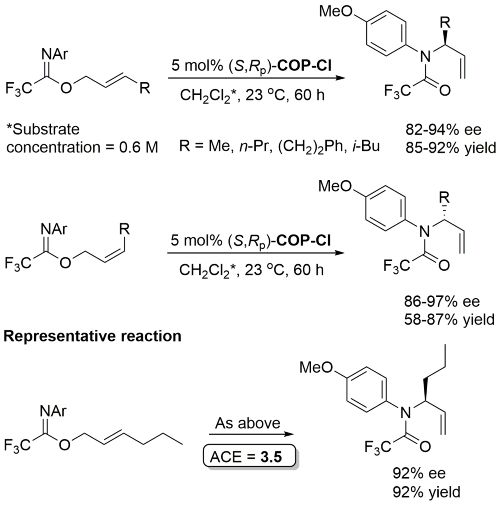
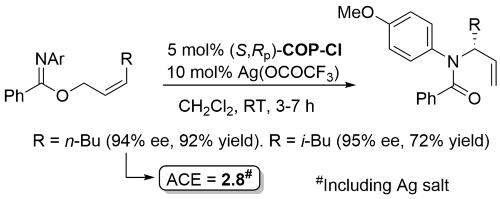
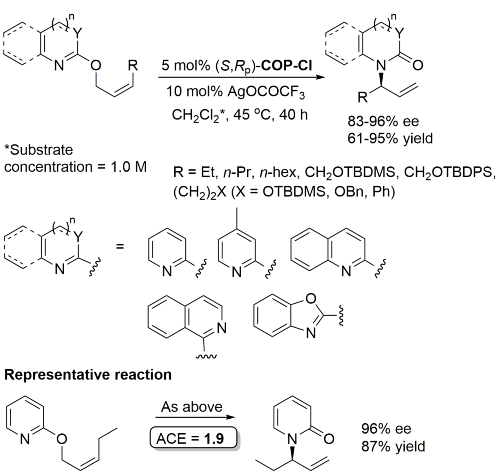
Palladacycle inactive as a catalyst without the addition of Ag(OCOCF3). Corresponding (E)-substrates reacted at a slower rate and reduced selectivity (45-50% ee). Substrate concentration not given.
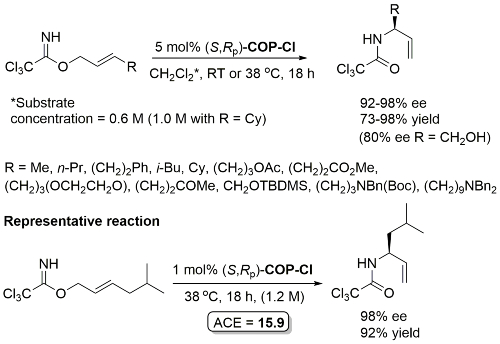
These conditions applied with R = (CH2)3OTBS in JACS11-133-18194 (e.e. not given). The structure of ‘(R)-COP-Cl’ (i.e. (R,Sp)-COP-Cl) is incorrectly drawn in this paper.
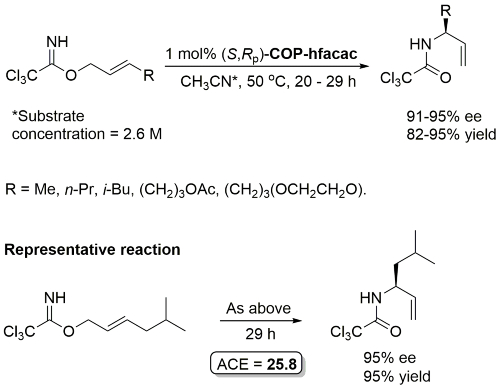
Unlike COP-Cl, COP-hfacac is soluble in a variety of solvents including THF, cyclohexane and toluene. Enantioselectivity essentially invariant of solvent, but impractically slow in cyclohexane or toluene (which also must be anhydrous).
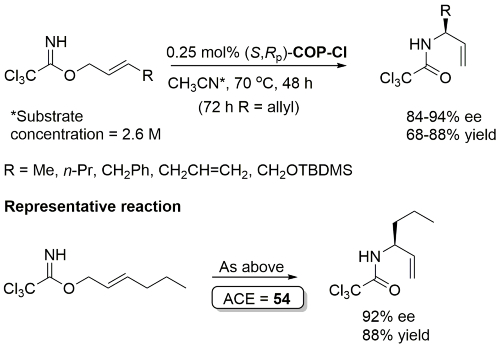
Same conditions but with 0.75 mol% catalyst applied to substrate with R = p-MeOC6H4CH2 (91% ee, 89% yield) in OL09-11-2892.
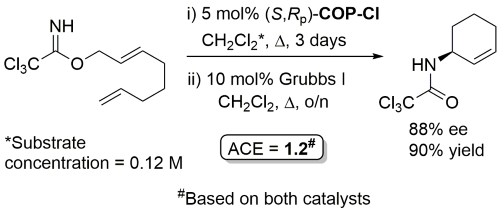
Method from OBC paper.
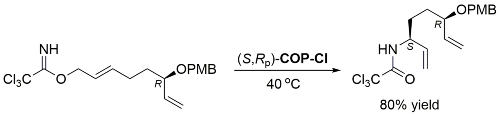
Catalyst loading, time, concentration and product diastereomeric ratio not given. With (S,Rp)-COP-Cl the S enantiomer of the starting material gave the S,S product, so under essentially complete catalyst control.

Ring-closing metathesis with Grubbs I followed by Kharasch cyclisation.
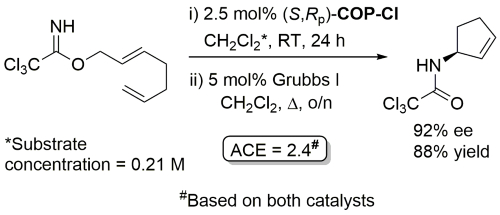
Yield based on precursor alcohol to trichloroacetimidate substrate. Multigram scale. The structure of “(R)-COP-Cl” is incorrectly drawn in the manuscript.
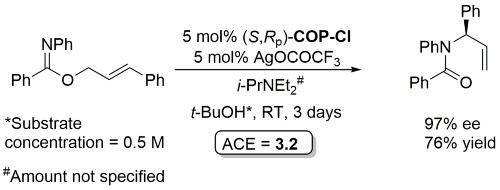
Rearrangement product incorrectly drawn as the S enantiomer in this paper. Correct R enantiomer drawn above.
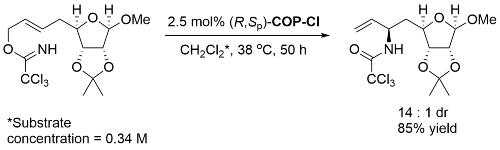
Use of the same substrate with (S,Rp)-COP-Cl gave the epimeric product with a dr of 29 :1 indicating predominantly catalyst control. The structure of “(R)-COP-Cl” is incorrectly drawn in the manuscript.
Pd – Rearrangement of allyloxy iminodiazaphospholidines JACS05-127-14887
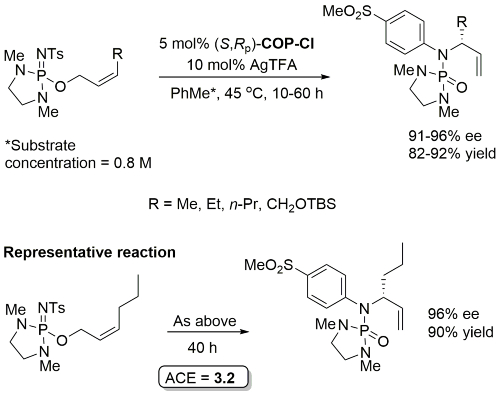
Initial rate of Z substrate (R = Et) estimated to be 14 times faster than corresponding E substrate. Lower ee values with corresponding E substrates [84-86%, S product with (S,Rp)-COP-Cl]
Pd – Intramolecular aminopalladation JOC05-70-2859
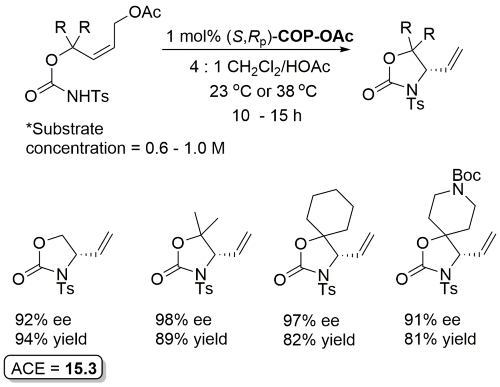
Tosylcarbamate substrates prepared from corresponding allylic alcohol and p-toluenesulfonyl isocyanate.
Pd – Intramolecular allylic substitution JOC10-75-7897
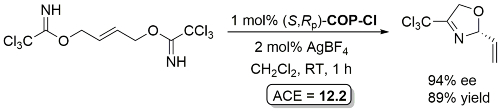
No specific experimental for this reaction – quantity of solvent used not given.
Pd – [3,3]-Sigmatropic rearrangement of allyloxy substituted N-heterocycles OL10-12-260
Pd – O-Allyl Carbamothioate Rearrangement OL08-10-1485
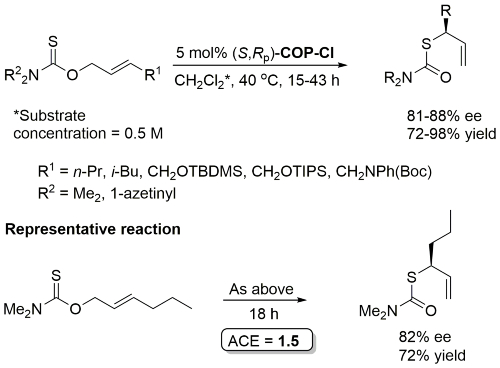
Reaction also performed at RT (67 h, 5 mol% with R1 = n-Pr, R2 = 1-azetinyl, 83% ee) and with 1 mol% cat (47 h, 40 oC, R1 = n-Pr, R2 = 1-azetinyl, 76% ee).
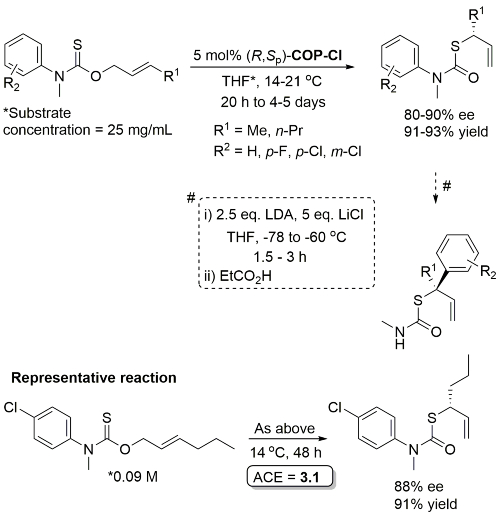
Higher ee with R1 = n-Pr than R1 = Me (for which several examples with <80% ee). Stereospecific N to C aryl migration (#) in some instances resulted in an erosion of ee (slow reaction relative to allyllithium racemisation).
Pd – Allylic Ester Synthesis JACS10-132-15185 (communication JACS05-127-2866)
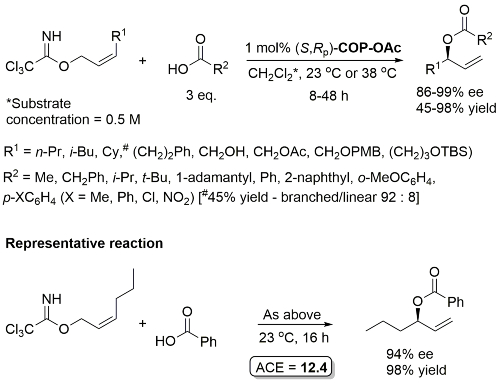
Much lower yield (and reduced e.e.) with COP-Cl. Lower e.e. with (E)-trichloroacetimidates and also competative allylic imidate rearrangement. Reaction temperature of 38 oC with bulkier (slower) carboxylic acids and/or those with lower solubilities. Complete (branched) regioselectivity excepting #. Mechanistic investigations and a model for the basis of enentioselectivity is reported in JACS10-132-15192.
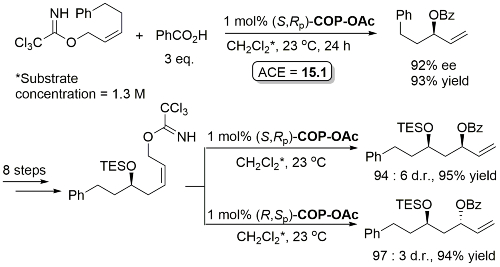
Iterative methodology also extended to the synthesis of all four protected triol enantiomers containing three stereogenic centres. All reactions show essentially complete catalyst control. Methodology further demonstrated by the total synthesis of (+)-solistatin.

Full paper version of CC07-4164. The butanoic acid nucleophiles provide a method for product ester deprotection (X = OTBS with TBAF, X = N3 with PPh3 then H2O, X = OPMB with DDQ then KOt-Bu), methodology applicable to orthogonal deprotection if other ester groups present.
Pd – Allylic Aryl Ether Synthesis OL07-9-911
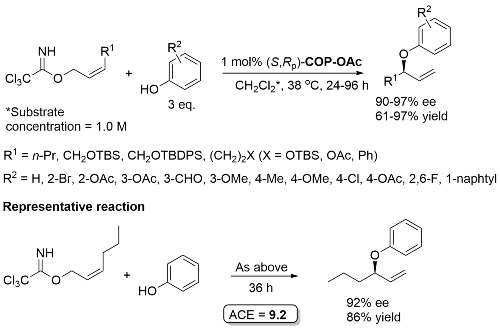
COP-Cl as catalyst gives comparable ee but inferior rate. One example with a branched γ-substituent with R1 = Cy (R2 = 4-OAc, 90% ee, 30% yield, 96 h). R2 = NO2, CN and allyl did not result in product formation.
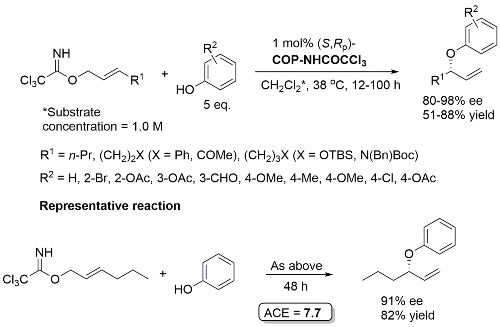
COP-NHCOCCl3 prepared from COP-OAc and 2,2,2-trichloroacetamide in benzene at 80 oC (azeotropic removal of acetic acid). Use of COP-OAc in the representative reaction resulted in a 59:41 ratio of phenyl ether (91% ee) to allylic imidate rearrangement product. With COP-NHCOCCl3 the corresponding ratio is 92:8.
Pd – Enantioselective synthesis of 2-vinyl oxygen heterocycles JOC12-77-1961
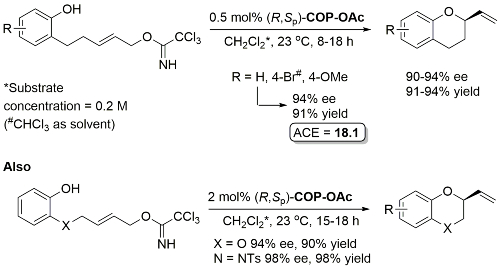
Use of COP-Cl instead of COP-OAc resulted in competitive allylic imidate rearrangement. A very low ee resulted from a corresponding Z substrate. Acetates rather than trichloroacetimidates may be used for the synthesis of 2-vinylchromanes resulting in similar enantioselectivities although with a higher catalyst loading (2 mol%) and with 1 eq. of KF as additive.
Pd – Transcyclopalladation JACS05-127-2388