Conjugate Grignard addition [Cu]
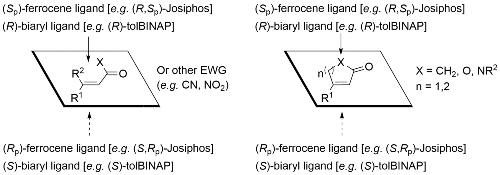
The following mnemonics for acyclic and cyclic substrates are applicable to copper-catalysed conjugate reduction, conjugate borylation and conjugate Grignard addition.
The catalytic cycle for conjugate reduction proceeds via in situ formation of a copper hydride species. This is tolerant to alcohols and water, and addition of the former significantly enhances the rate of catalyst turnover. This effect is ascribed to faster quenching of the initially formed copper enolate by the alcohol followed by σ-bond metathesis of the resulting copper alkoxide with the silane to regenerate L*Cu-H. Use of a bulky alcohols such as t-BuOH reduces the rate of the reaction of the alcohol with the silane resulting in the formation of H2.
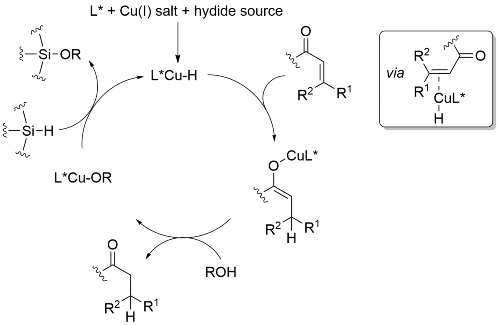
A corresponding catalytic cycle accounts for conjugate borylation with L*Cu-H replaced by L*Cu-Bpin, with this generated from the reaction between L*Cu-OR and B2pin2. There is no need to employ a bulky alcohol ROH, and methanol is typically used. Similarly, L*Cu-H is replaced by L*Cu-R for Grignard conjugate addition. Fortunately, in this alcohol free reaction, the rate of transmetallation between the Grignard reagent and the copper enolate is sufficiently high to allow catalyst turnover.
Conjugate Reduction
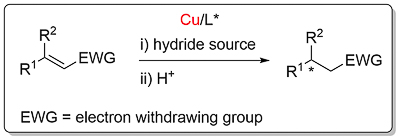
EWG = COR3 (A,B): CO2R3 (A): CN (A): NO2 (A): C=N (within heterocycle) (A). Additional examples employing non-ferrocene based ligands can be found here. More information here.
Conjugate Borylation
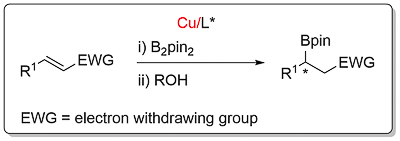
EWG = COR2 (A,B,C,D,E,F,G): CO2R2 (A,B,C,D,E,F,G,H): CN (A): C=N (imine) (A): CONR2 (A): SO2Ar (A): P(O)Ph2 (A). More information here.
Conjugate Grignard Addition
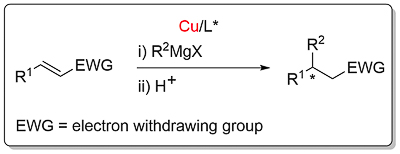
EWG = COR2 (A, B, C, D, E, F, G), chromene COR2 (A): CO2R2 (A, B, C, D), pyranone CO2R2 (A): COSR2 (A, B, C, D): C=N (within heterocycle) (A): CONR2 (A). More information here.
Ligand Availability
p-tolBINAP
Aldrich (S) (R) Strem (S) (R) TCI (S) (R)
DTBM-SEGPHOS
Aldrich (S) (R) Strem (S) (R) TCI (S) (R)
SYNPHOS
Additional conjugate reduction examples:
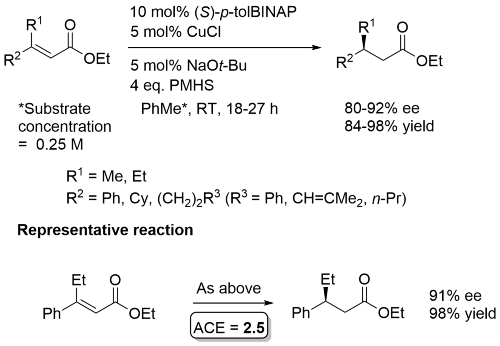
Changing the substrate configuration results in the opposite enantiomer. For the representative example, changing to the (Z)-isomer gave the R reduction product in 83% ee (96% yield).
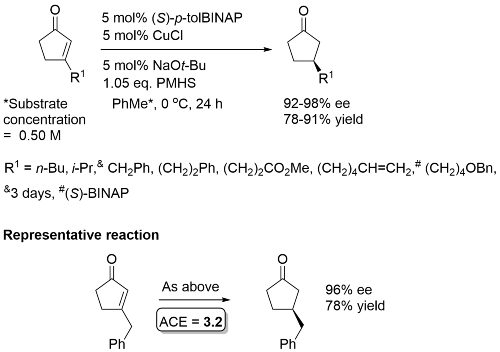
Substrate with R1 = Me reduced at -78 oC (94% ee). If larger amounts of PMHS used over reduction to the saturated alcohol observed. No reduction for R1 = t-Bu, and with 2,3-disubstituted substrates. Cyclohexenones (R1 = Me [at -78 oC], n-Bu) and a cycloheptenone (R1 = (CH2)2Ph) reduced under the same conditions with a longer reaction time, with same sense of enantioselection, excepting that (S)-BIPHEMP used for the 6-ring, and (S)-BINAP for the 7-ring.
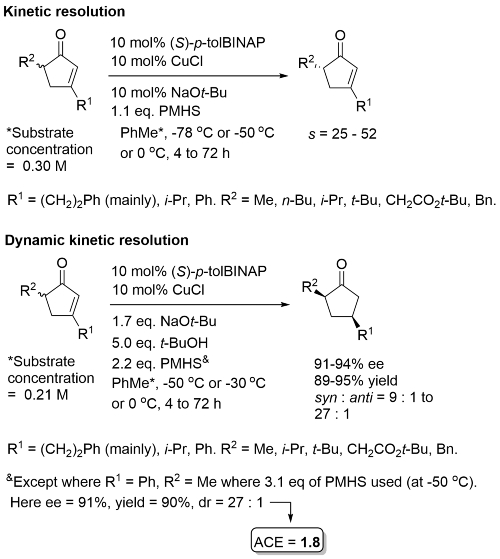
For R2 = Me or primary alkyl a reaction temp of -78 oC (KR) or -50 oC (DKR) employed. Postulated that DKR is possible because the initially formed copper enolate arising from L*CuH reduction undegoes σ-bond metathesis with the silane to give a silyl enol ether product (and L*CuH) that cannot epimerise. TBAF employed in the work-up. 5 eq. of t-BuOH required for a usable rate of racemisation. Kinetic resolution of 3,4-dialkylcyclopentenones gave lower s (<10).
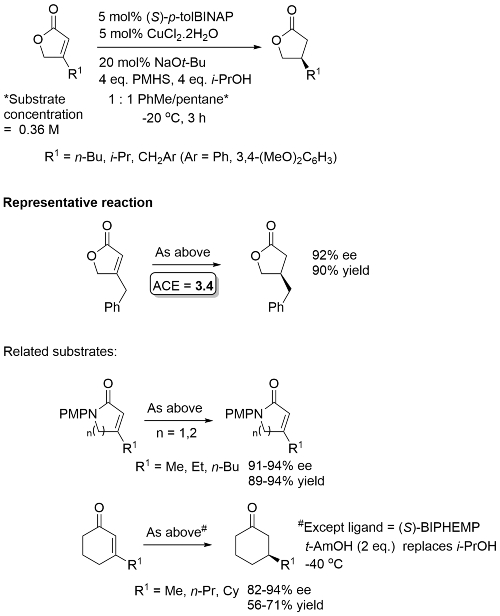
NH and NBn lactams did not undergo reduction. [Ligands used here = (S)-p-tolBINAP, (R)-p-tolBINAP, (S) and (R)-BIPHEP ((6,6′-Dimethyl-[1,1′-biphenyl]-2,2′-diyl)bis(diphenylphosphine) not available from major catalogue suppliers].
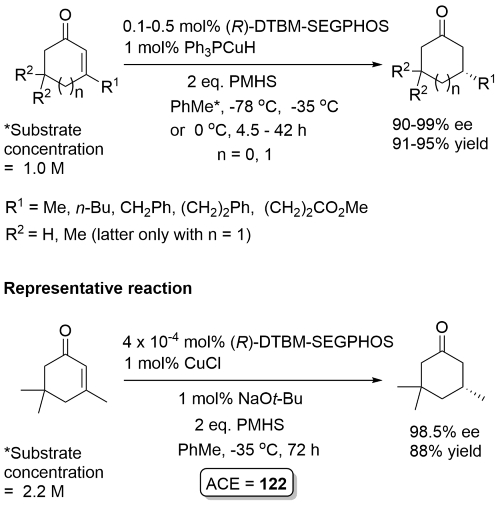
Accelerated rate with DTBM-SEGPHOS [(R) and (S) enantiomers commercially available] compared to p-tol-BINAP or BIPHEMP. No over reduction using 2 equivalents of PMHS. For the substrate used in the reprentative reaction higher ee values obtained in THF and dioxane/THF compared to toluene. Viable to replace NaOt-Bu with NaOMe.
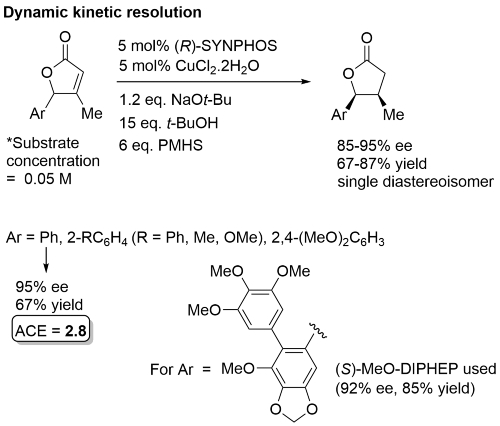
NOTE – This paper is vague on the absolute configuration of the products of DKR. The implication is as given in the scheme above, BUT (R)-SYNPHOS [(S)-SYNPHOS also available] is expected to give the enantiomeric product. Absolute configuration in the scheme above correct for the expected product obtained from (S)-MeO-BIPHEP [(R)-MeO-BIPHEP] Substrates that contained an alkyl rather than an aryl substituent at the γ-position did not undergo DKR (no more than 50% conversion), and the product was a mixture of diastereoisomers with <25% ee for the cis product.