Cobalt oxazoline palladacycle [Pd]
Iridium/Phosphoramidite Catalysed Allylic substitution [Ir]
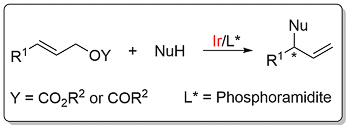
Key aspects of this methodology are given below. Applicable to amination, alkylation, etherication, hydroxylation, sulfidation, sulfination, sulfonation.
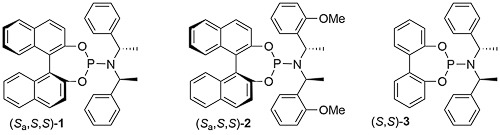
A key breakthrough in the use of iridium in enantioselective allylic substitution reactions was the discovery that Feringa-type ligands such as 1 work very well [JACS02-124-15164, JACS03-125-14272]. The initially formed complex A, resulting from the addition of 1 to [Ir(COD)Cl]2, is not the catalyst. Instead formation of an iridacycle by methyl C-H group activation results in B, dissociation of L resulting in the catalyst C. As a result, these reactions need to be run under basic conditions.
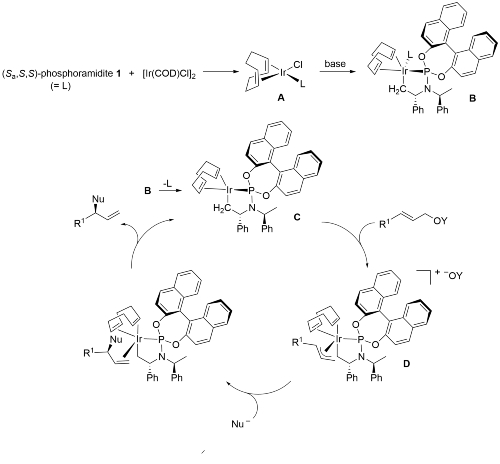
The allyliridium intermediate D may be synthesised from the ligand, [Ir(COD)Cl]2 and an allylic carbonate (with the counter ion X- introduced from AgX – CEJ09-15-11087). The complexes are air-stable and may be purified by column chromatography. Just one, the thermodynamically most stable, of the 16 possible stereoisomers is formed. Replacement of COD with dbcot gives a catalyst that operates under aerobic conditions, and results in an improvement in enantioselectivity and regioselectivity [Angew08-120-7652].
Use of a related achiral catalyst with an enantiomerically pure substrate revealed a conservation of enantiomeric purity [CC99-741]. This stereospecificity reveals that the rate of nucleophilic attack is faster than the rate of isomerisation of the intermediate allyl complex. Oxidative addition and attack of the nucleophile both proceed with inversion of configuration, resulting in overall retention of configuration. By extension, with a prochiral substrate the stereo-determining step is oxidative addition, and use of racemic branched allylic substrates results in low enantioselectivity.
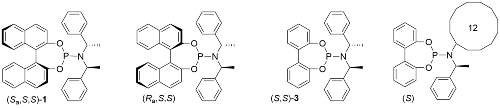
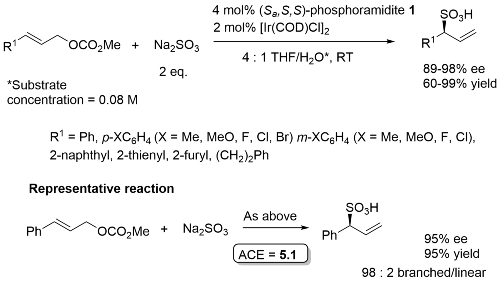
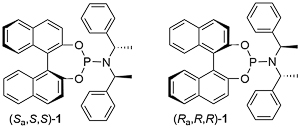
Use of ligand (Sa,S,S)-1 results in a much faster reaction compared to the use of its (Ra,S,S) diastereoisomer [JACS02-124-15164]. This is accounted for, at least in part, by the much faster rate of cyclometallation of (Sa,S,S)-1 compared its (Ra,S,S) diastereoisomer [JACS05-127-15506]. In addition, (Sa,S,S)-1 is the matched ligand, and the (Ra,S,S) form the mismatched ligand, with respect to product enantioselectivity [see also EJOC03-1097].
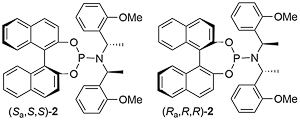
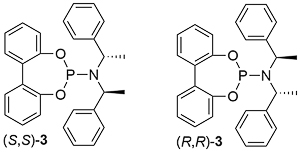
Replacement of the BINOL derived moiety by 2,2’-biphenol to give ligand (S,S)-3 resulted in a modest reduction in enantioselectivity [Angew04-43-2426] (95% ee for 1 vs. 87% ee for 3 in a representative allylic amination). A reduction in the number of chirality elements to just one (i.e. this being the stereogenic centre within the resulting iridacycle ring) was achieved with a cyclododecane containing ligand, this resulting in ee values of >90% ee for allylic amination reactions [JACS05-127-15506].
Review: CR19-119-1855. The following illustrates a number of representative reactions (and is very far from exhaustive).
Allylic amination

Phosphoramidite 1. Excellent regioselectivity (branched : linear typically about 50 : 1, lowest = 11 : 1). With R2 = H typically small quantity of double alkylation (<5%). Lower regioselectivity (6 : 1) with R1 = p-NO2C6H4 (86% ee) and ee = 76% with R1 = o-MeOC6H4.
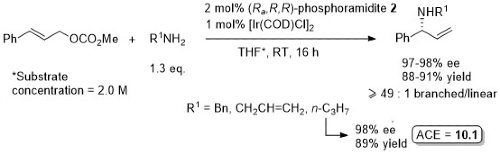
Phosphoramidite 2. Significant acceleration in the reaction rate noted with phosphoramidite 2 compared to phosphoramidite 1.
Allylic alkylation
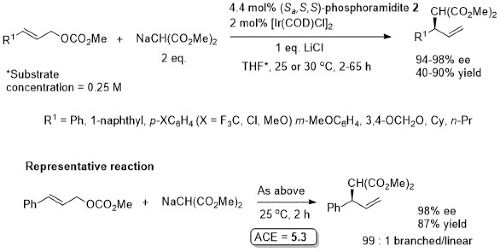
Phosphoramidite 2. Most R1 substituents gave ≥99 : 1 branched/linear. Lower ration with R1 = Cy (13 : 1) and R1 = n-Pr (4 :1). With R1 = o-MeOC6H4, ee = 79%.
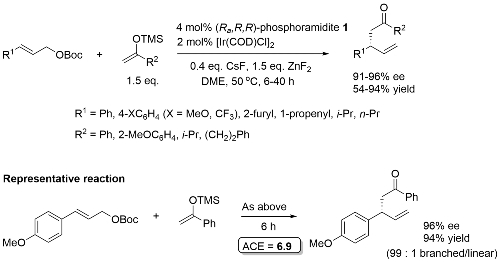
Phosphoramidite 1. Solvent concentration not given. Somewhat lower branched/linear ratio (ca. 6 : 1) where R1 = i-Pr, n-Pr. Both CsF and ZnF2 needed – no reaction with ZnF2 only. Substoichiometric CsF aids avoidance of diaallylation.
Allylic etherication
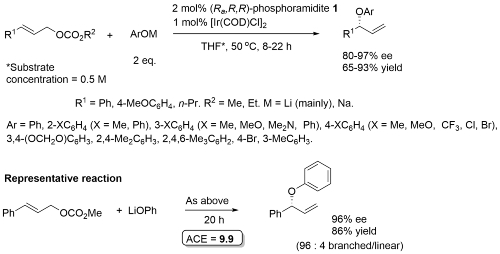
Phosphoramidite 1. Excellent regioselectivity (branched : linear typically about 25 : 1, lowest = 7 : 1). With R1 = 2-MeOC6H4 (R2 = Me, Ar = Ph) ee = 75%. Lithium phenolates prepared from the reaction of the corresponding phenol with n-BuLi.
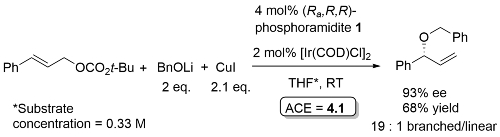
This chemistry was exemplified using the derivative of phosphoramidite 1 containing two 1-naphthyl groups rather than two phenyl groups [Ra,R,R] [Sa,S,S] – 2 mol% ligand, 94% ee, 91% yield, 99:1 branched/linear. Used double the catalyst loading with sec and tert-alkoxides (and longer reaction times). Copper (and zinc) alkoxides required (no reaction with lithium alkoxide). Tert-butyl ester to avoid transestrification. Use of both enantiomers of chiral sec-alkoxides resulted in essentially one or the other product diastereoisomers (i.e. no matched/mismatched influences).
Allylic hydroxylation
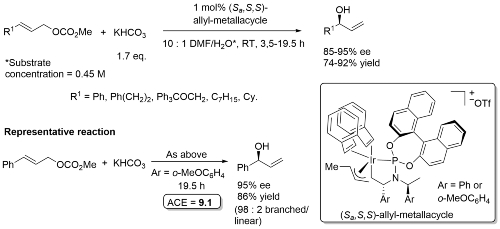
The iridacycle catalyst is derived from either Phosphoramidite 1 or Phosphoramidite 2 and dbcot as described in the supporting information. Higher e.e. values obtained with Ar = o-MeC6H4 (approx 5% higher). These iridacycles catalysts are stable to air and water – by extension, no useful results were obtained with a corresponding COD-containing catalyst. The bicarbonate anion is thought to be the nucleophile, followed by decarboxylation.
Allylic sulfidation
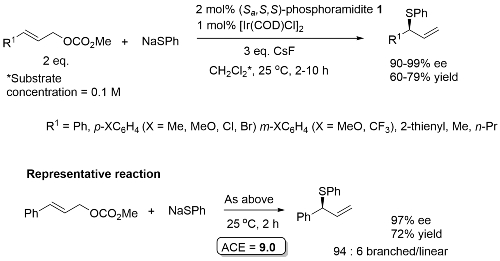
Allylic sulfination
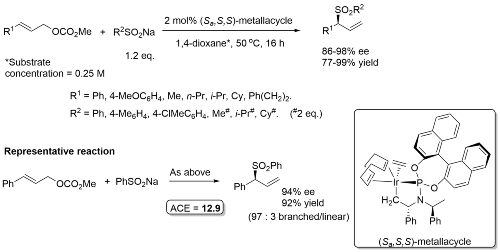
The iridacycle catalyst (not commercially available) contains a labile ethene ligand, loss of which gives a 16-electron species with which the catalytic cycle is initiated by oxidative addition (see JACS09-131-8971). Generally excellent branched to linear selectivity (≥99 : 1).
Allylic sulfonation
Phosphoramidite 1. No reaction times given.
Ligand Availability
Phosphoramidite 1.
Aldrich (Sa,S,S) Strem (Sa,S,S) TCI (Sa,S,S) (Ra,R,R)
Phosphoramidite 2.
Aldrich (R,R) Strem (S,S) (R,R) [011123]
Cobalt oxazoline palladacycle Catalysed Allylic Substitution [Pd]
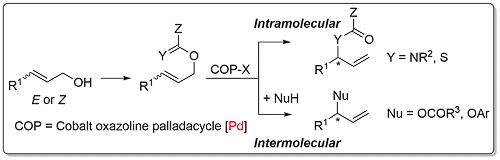
COP-X catalysts are described here. Links as Y = NR2 (A, B, C), Y = S (A), Nu = OCOR3 (A), OAr (A, B). Key aspects of this methodology are as follows.
The allylic imidate rearrangement, also known as the aza-Claisen or Overman rearrangement, is an example of a [3,3]-sigmatropic rearrangement. Allylic alcohol derived trichloroacetimidates are especially useful for this reaction due to the ease of synthesis (from trichloroacetonitrile), and the product of the rearrangement is easy to hydrolyse resulting in the generation of an allylic amine. The reaction is essentially irreversible due to the strong driving force for the rearrangement step (ΔH = -70 kJ mol-1), but in the absence of a catalyst the concerted reaction requires a high temperature.
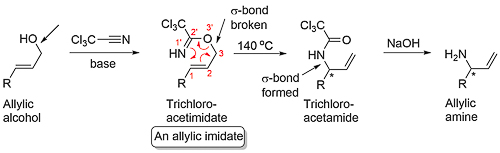
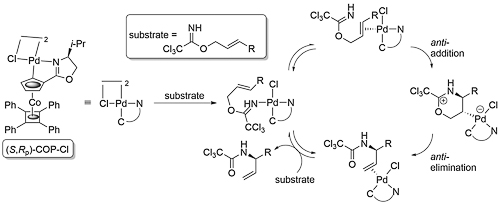
A key breakthrough was the discovery that the reaction is catalysed by Hg(II) and Pd(II) salts, with rate accelerations of 1010 or more (Angew84-23-579). The mechanism for the stepwise metal catalysed reaction involves stereospecific anti-addition (iminopalladation) to a metal-activated C=C bond, followed by the resulting intermediate undergoing stereospecific anti-elimination (deoxypalladation).
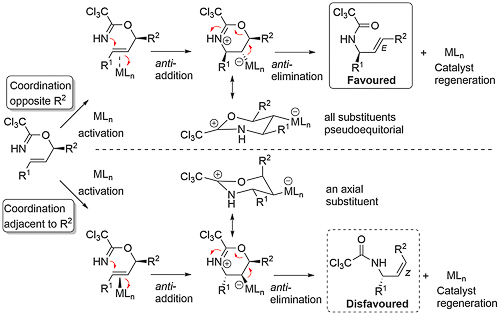
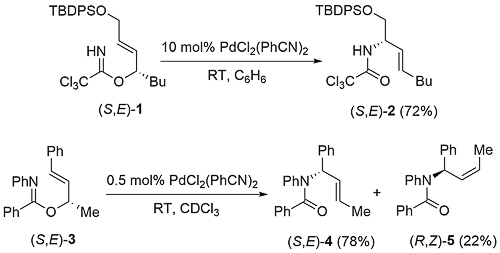
For a chiral substrate (R2 ≠ H), anti-addition via η2-coordination of the metal opposite the R2 substituent gives a six-membered cyclic intermediate with pseudo-equatorial substituents, with subsequent anti-elimination leading to an (E)-allylic amide. Alternatively, reaction via η2-coordination of the metal adjacent to the R2 substituent gives an intermediate with a pseudo-axial substituent, leading in turn to a (Z)-allylic amide. The diastereoselective preference for the first of these options is seen with the exclusive conversion of (S,E)-trichloroacetimidate 1 into (S,E)-amide 2 (TL92-33-4313) In contrast, N-phenyl benzimidate (S,E)-3 reacts a modest diastereoselectivity to give (S,E)-4 and (R,Z)-5 (3.5 : 1) (JACS85-107-2058). The completely E selective rearrangement of other chiral trichloroacetimidates has also been reported (Tet92-48-1071).
The following catalytic cycle for COP-Cl catalysed allylic imidate rearrangement has been proposed (JACS07-129-5031).
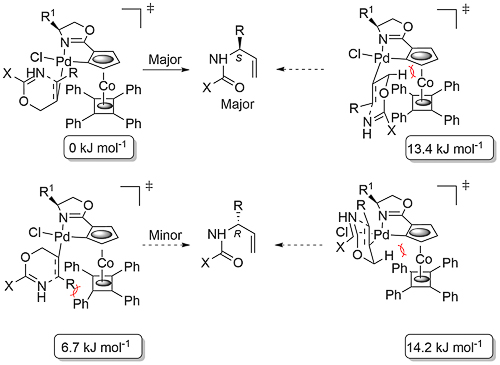
DFT calculations reveal a significantly lower transition state energy for the anti-addition step with the double bond coordinated trans rather than cis to the oxazoline nitrogen (which is less electron donating than the palladacycle carbon). Calculations and kinetic measurements further reveal that the rate and enantioselectivity determining step is anti-addition. These outcomes led to the relative energy determination of the following four possible transition states where X = H, R = R1 = Me. The lowest in energy corresponds to the observed (S)-configuration for the major enantiomer, and the difference to the next lowest [giving (R)] is 6.7 kJ mol-1, which would result in a 15:1 (88% ee) selectivity at 25 oC, very close to the actual selectivity observed with X = CCl3, R = Me, R1 = i-Pr [89% ee (CEJ07-13-10216) and 92% ee (JACS03-125-12412)].
Trichloroacetimidates of (Z)-configuration are less prone to undergo this rearrangement, such that in the presence of a carboxylate or phenolate nucleophile an alternative SN2’ substitution is observed. COP-OAc is significantly more active as a catalyst than COP-Cl; the greater lability of the acetate ligand compared to a chloride ligand aids the formation of the substrate chelated cationic intermediate A of the following proposed catalytic cycle (JACS10-132-15192).
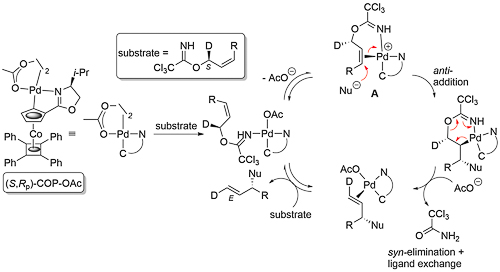
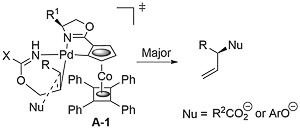
That the reaction proceeds by an anti-addition/syn-elimination pathway is supported by the transformation of an S deuterated precursor into the E product with (S,Rp)-COP-OAc (the catalyst controlling the facial selectivity). That A contains an alkene trans to nitrogen in the rate and enantioselectivity determining nucleophile addition step was supported by calculations, and of the two options with opposite facial selectivity, transition state A-1 is lower in energy (by 6.3 kJ mol-1, which would result in a 12.6 : 1 (85% ee) selectivity at 25 oC).
Pd/Ligand [Pd]
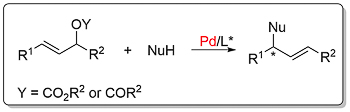
Numerous ligands have been applied to this reaction including ferrocene-based examples (where generally R1 = R2). Links as: alkylation (A, B, C), amination, phosphination, phosphonation.