Other acronyms used for these ligands include FOXAP, FcPHOX and DIPOF. First reported by Richards and Uemura, the synthesis of these ligands was improved by utilisation of the highly diastereoselective lithiation conditions reported by Sammakia. The following lists reactions, organised by metal (Pd Ru Ir Cu Ag Ni), for which the application of these ligands has resulted in >80% ee. ACE = Asymmetric Catalytic Efficiency. Literature covered until end 2018 (71 entries – including [Phosferrox-Ru].
Palladium
Pd – Heck TL99-40-9163. TL00-41-2261. JMCA03-196-65.
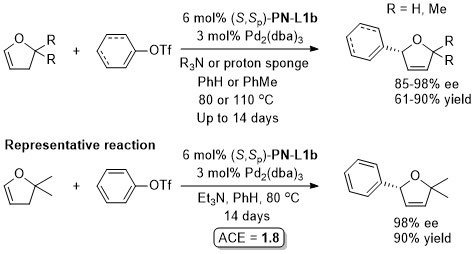
Pd – Alkylative ring opening JACS00-122-1804. OL00-2-1971. OL02-4-1879.
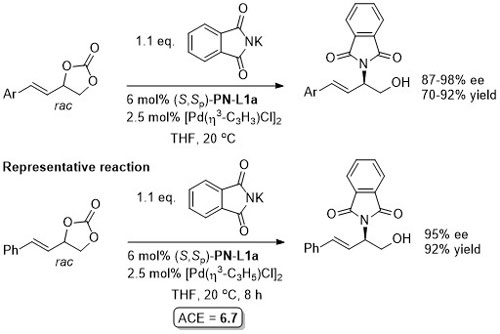
Pd – Allylic amination OBC13-11-7412.

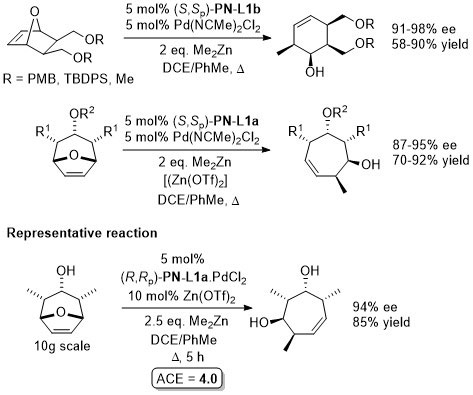
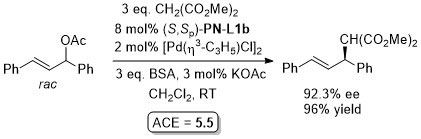
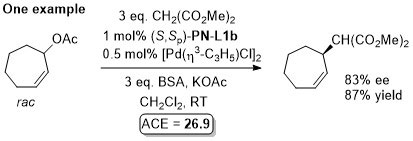
Pd – Allylic alkylation BCKS97-18-789. TA97-8-1179.
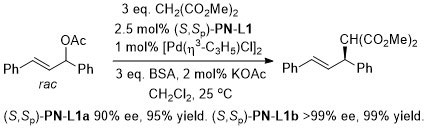
GC02-4-103. This reaction with (S,Sp)-PN-L1a in ionic liquid [bmim][PF6] (up to 86% ee)

Pd – Intramolecular arylation JACS17-139-16486
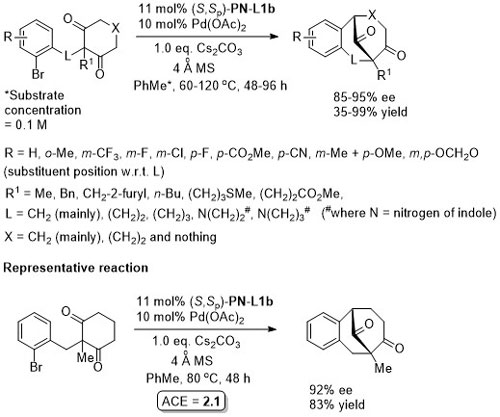
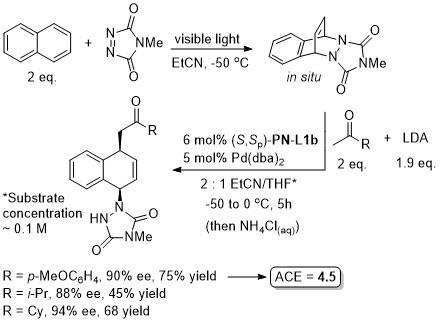
Pd – Diazabicycle desymmetrisation JACS17-139-17787
Pd – 3 + 2 cycloaddition ACSCat16-6-6408
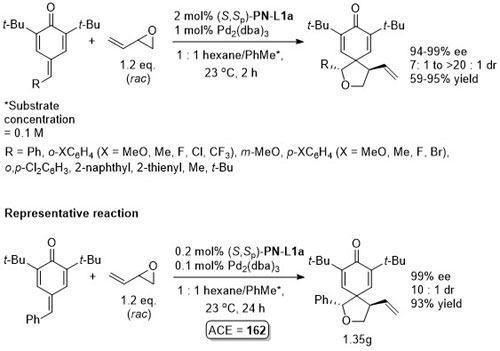
One example where the t-Bu quinone substituents are replaced by i-Pr substituents but this resulted in a signifcant reduction in ee (to 74% with R = Ph).
Pd – Imine arylation OCF2019-6-1572
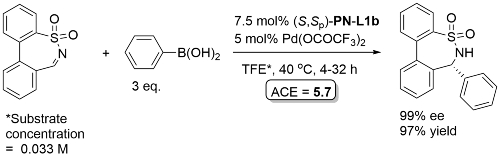
Single example from a ligand scoping study. Reaction subsequently exemplified with (S)-t-Bu-PHOX ligand, and absolute configuration of the product assigned by analogy.
Ruthenium
Ru – Transfer hydrogenation OL97-62-6104
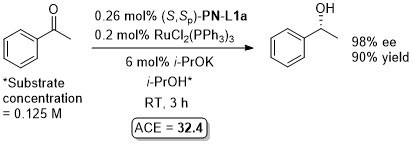
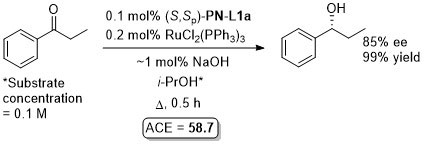
Similar results from in situ generated catalyst also reported in JOMC99-572-163. This paper also describes the preparation of [(S,Sp)-PN-L1a]RuCl2(PPh3). An octahedral adduct derived from this, [(S,Sp)-PN-L1a]RuCl2P(OMe3)2, has also been applied to transfer hydrogenation (94% ee, 96% conversion from PhCOMe, ACE = 71.8), see: JOMC08-693-2535.
Iridium
Ir – Hydrogenation ASC04-346-909
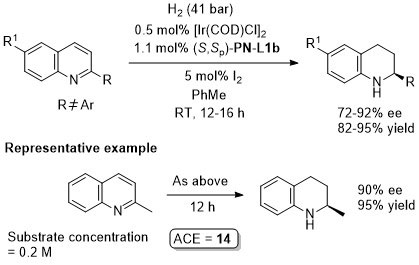
TA07-18-629 The catalyst in this example is the isolated adduct derived from (S,Sp)-PN-L1b, [Ir(COD)Cl]2 and NaBArF.
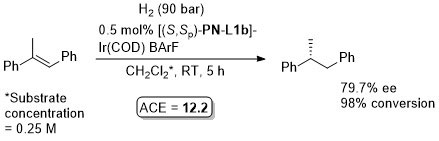
Ir – Hydrosilylation Orgmet99-18-2271
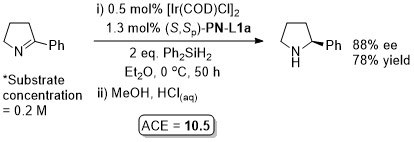
Copper
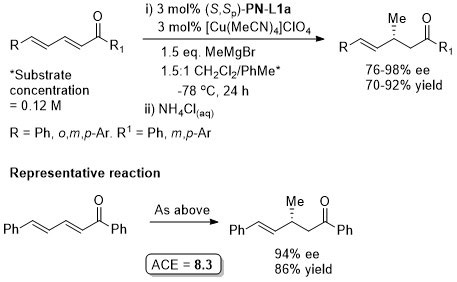
Cu – Conjugate addition Tet97-53-16503
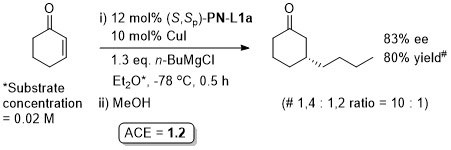
OL10-12-1080 See corrigendum for correct assignment of absolute configuration.
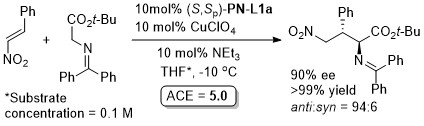
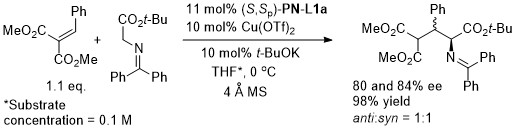
With the 3,5-(CF3)2C6H3 analogue of (S,Sp)-PN-L1a 8 : 1 anti : syn diastereoselectivity resulted.
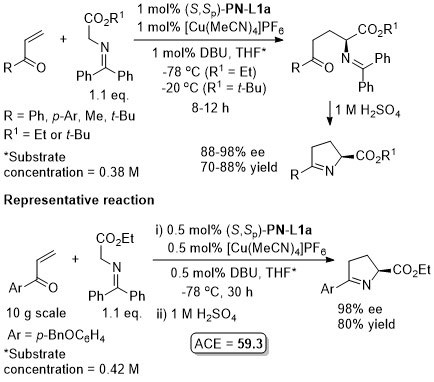
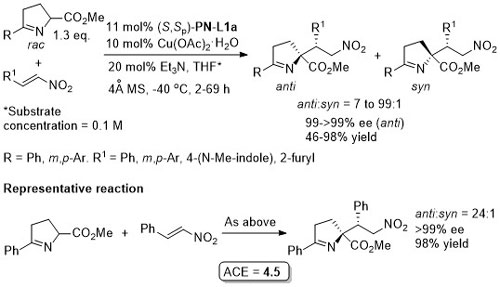
Lower anti:syn selectivity with R1 = o-Ar, t-Bu and CH2OBn. High syn selectivity with all R1 substituents achieved using a 2,3-dihydroimidazo[1,2-a]pyridine-OH (i.e. an N,O-ligand) under the same conditions at RT.
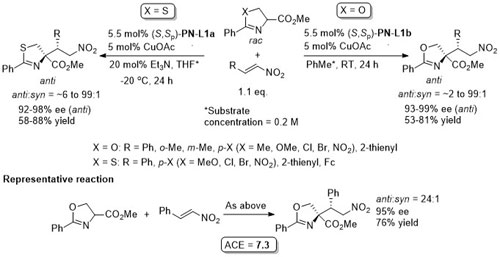
Anti selectivity [(4R, 1’S) using (S,Sp)-PN-L1a] changes to syn (>15: 1)selectivity (4R, 1’R) where the Ph groups of (S,Sp)-PN-L1a are replaced by (3,5-CF3)2C6H3 groups.

Addition of water* is required to suppress pyrrolidine formation as a result of 5-endo-trig cyclisation (!) of the enolate resulting from conjugate addition on the carbon of the diphenylimine moiety (the chemoselectivity refered to above). [*The addition of water is not included in the method given in the SI.] A later publication [ASC18-360-2144] reported that changing to PPFA or a MingPhos ligand (exemplified in detail) gave the [3 + 2] cycloaddition products under the same conditions.
Cu – Conjugate addition-elimination OL09-11-2073
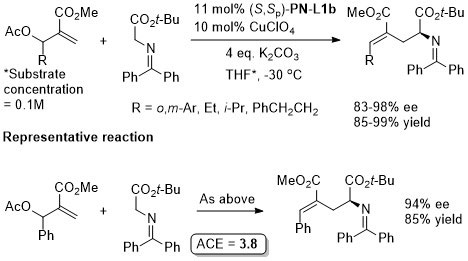
Cu – Conjugate addition/Friedel-Crafts cyclisation ACSCat16-6-5685

Conjugate addition chemistry closely related to similar Cu-based chemistry. See OL10-12-1080. In an intial ligand scoping study PN-L1a gave 97% ee vs. >99% with PN-L1c, and lower dr and chemoselectivity was observed with PN-L1a. The overall reaction is described as a [3+3] annulation. The alternative [3+2] cycloaddition pathway (see above for structure) was observed when the reaction was performed at RT. Cu(II) and Ag(I) salts also gave the [3 + 2] product, and this was observed with R1 = Cy under the standard Cu(I) conditions described above.
Cu – Mannich reaction JACS08-130-14362
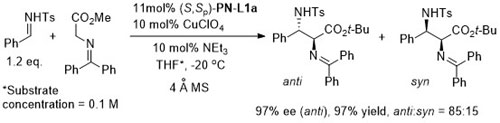
Higher anti selectivity with 4-MeOC6H4 analogue of (S,Sp)-PN-L1a. Reaction syn selective with the 3,5-(CF3)2C6H3 analogue of (S,Sp)-PN-L1a. In both cases ees generally >96%.
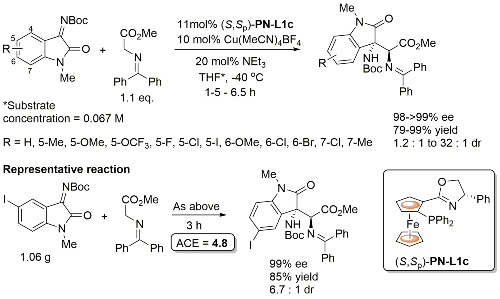
Initially developed with PN-L1a, PN-L1c gave similar enantioselectivity and higher diastereoselectivity. Where measured the minor diastereoisomer (epimeric w.r.t. the oxindole based stereogenic centre) also formed in high ee. Also one example with Boc replaced by Cbz (R = H, >99% ee, dr = 5 : 1).
Cu – 3 + 2 cycloaddition Angew06-45-1979
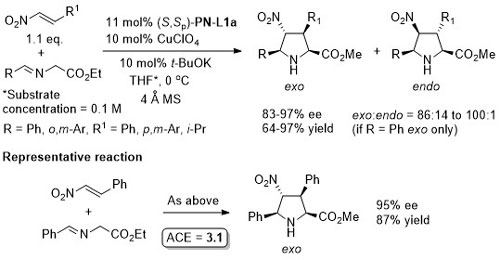
With the 3,5-(CF3)2C6H3 analogue of (S,Sp)-PN-L1a these reactions are endo selective.
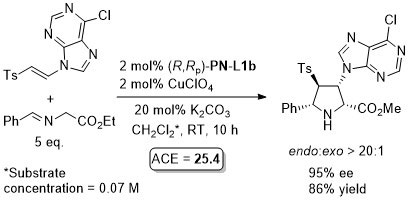
Reaction further exemplified with the (R,R,Rp)-Phosferrox ligand derived from (R,R)-2-amino-1,2-diphenylethanol. Major diasteroisomer described as endo, but should be described as exo.

One example with (S,Sp)-PN-L1a from a scoping study in which a Ming-Phos ligand gave a similar result (94% ee, 91% conversion, 15 : 1 dr at -40 oC). Further reaction optimisation and exemplification was with a Ming-phos ligand.
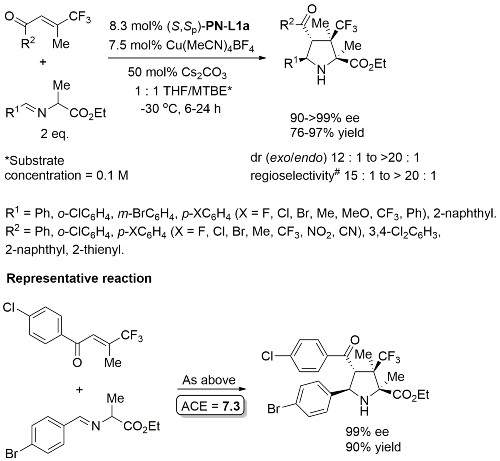
Methodology also applicable to a β-CHF2 substituent or a β-CH2F in place of CF3, but unsuccessful where CF3 replaced by CCl3 or Me. Use of (R,Sp)-PN-L2 under the same conditions resulted premoninantly in an alternative regioisomer (#). See entry under (R,Sp)-PN-L2. Rationalised as a result of differeing coordination modes, with PN-L1a = bidentate and PN-L2 = pseudobidentate.
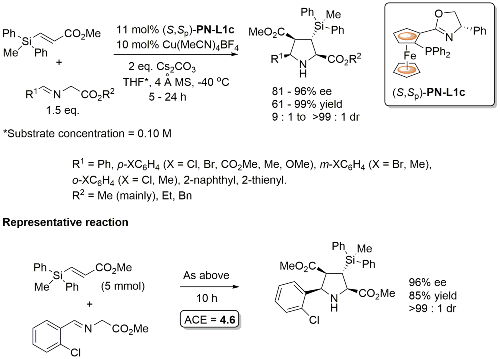
PN-L1c is not commercially available. Synthesis described in Orgmet95-14-5486. A scoping study gave an ee of 61% with PN-L1b and 87% with PN-L1c at RT. Use of PN-L1c and AgCO2CF3 at RT gave 73% ee. Little difference in enantioselectivity where Ph2MeSi replaced by PhMe2Si and Ph3Si. Major diastereoisomer rationalised by Ph2MeSi clash with Phosferrox PPh2 moiety in TS. Major diastereoisomer descibed as exo – note this relative stereochemistry has been described as endo previously (see Angew06-45-1979). Use of R1 = Cy (R2 = Me) resulted in poor dr (1. 4 : 1) but good ee for both diastereoisomers (>90%).
Cu – 4 + 2 cycloaddition Angew14-53-4680
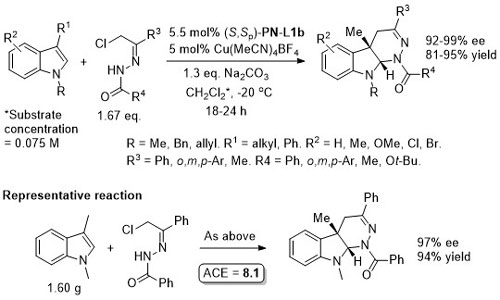
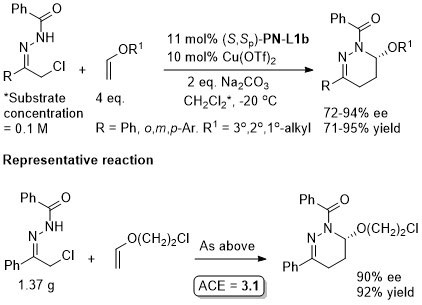
Cu – 3 + 3 cycloaddition Angew13-52-12377
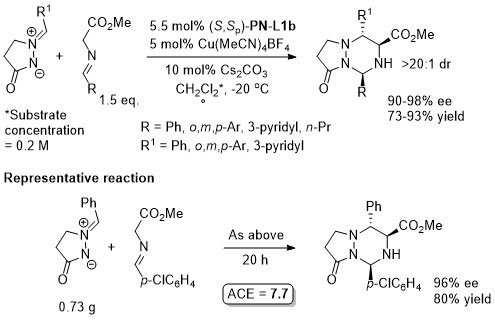
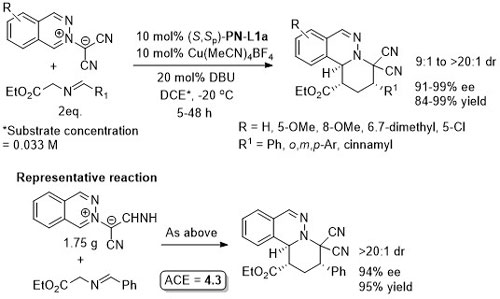
Cu – 3 + 4 cycloaddition ASC16-358-3748
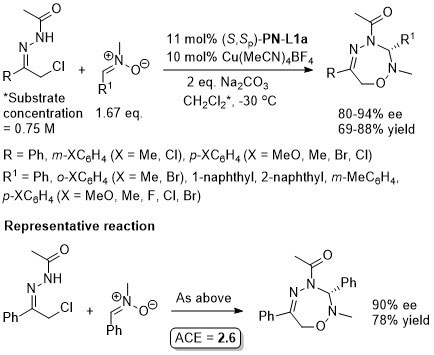
Cu – 3 + 6 cycloaddition OL15-17-1365
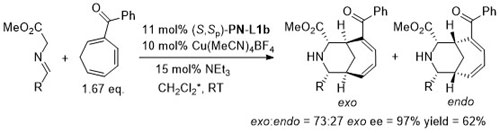
With N-methyl-1-[2-(diphenylphosphino)ferrocenyl]ethylamine >20:1 exo:endo was achieved (ee >95%).
Cu(Pd) – Allylation of α–amino acid derivatives Angew17-56-12312
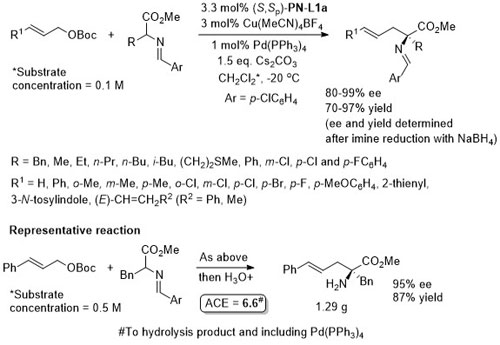
Also applied to the double allylation of a glycine derived aldime ester.
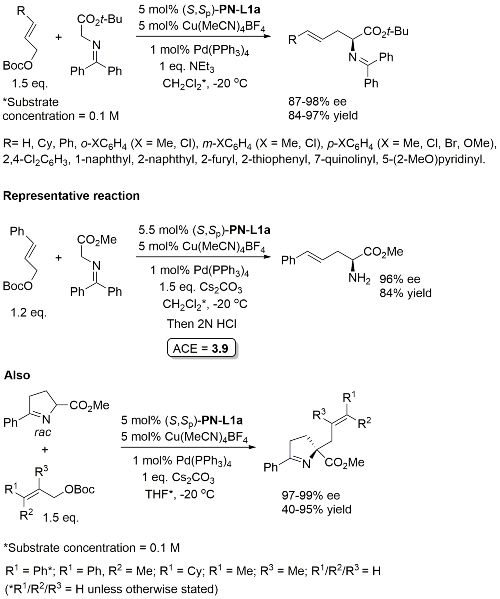
Catalyst loading quantities differ in paper and SI. Above values from SI. There appears to be no discussion of how the absolute configuration was determined, but the absolute stereochemistry given is in accord with previous literature. Glycine methyl ester derived ketimine used in a scale up reaction (the representative reaction) suggesting that a t-Bu ester is not needed for high ee. Use of the cyclic ketimine ester generally gave high linear/branced ratios, the exception being with R1 = Ph with R2/R3 = H.
Cu(Pd) – Allylation of pyrroline derivatives OL18-20-6564
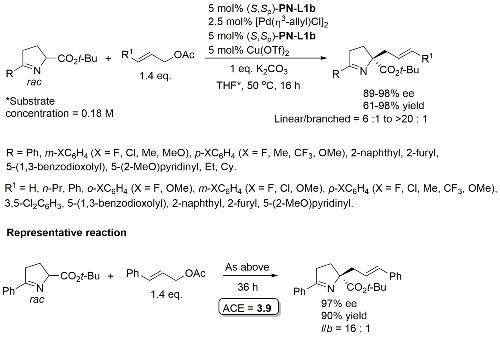
Tert-butyl ester required for high linear/branched ratio. Typically in the order of 1 : 1.5 (i.e. branched favoured – low dr) with corresponding methyl esters. Also one example of the use of 1,3-diphenylallyl acetate (dr = 7 : 1, >99% ee).
Cu(Ir) – Allylation of α–amino acid derivatives JACS18-140-1508
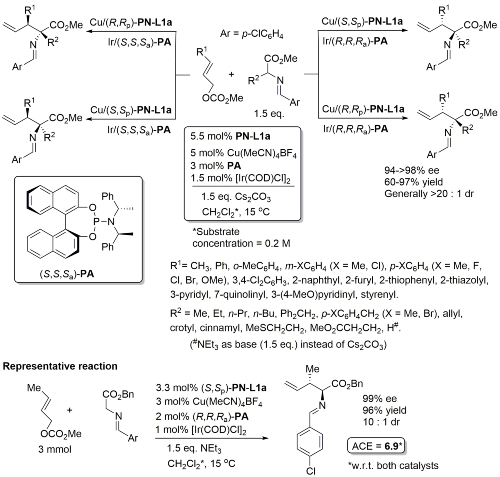
Details of (S,S,Sa)-PA here. Results above obtained from experiments where the two metal/ligand complexes were prepared separately to avoid potential ligand scrambling. Subsequent experiments showed that a one-pot protocol (i.e. combining together Cu(MeCN)4BF4, [Ir(COD)Cl]2, PN-L1a and PA] also works well (due to formation of an iridacycle as the actual Ir-based catalyst from PA only).
Cu – Beta (1,4) borylation OL14-16-1426
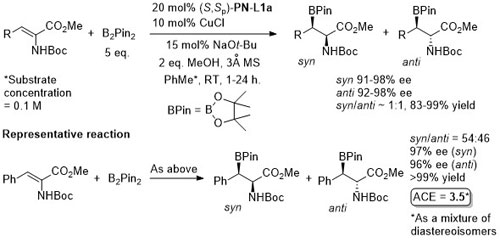
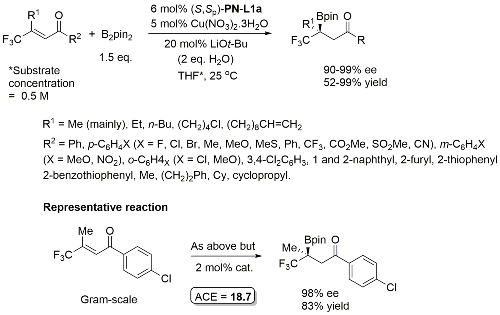
Cu – Sulfonylindole substitution (via in situ vinylogous imine) OL10-12-1688
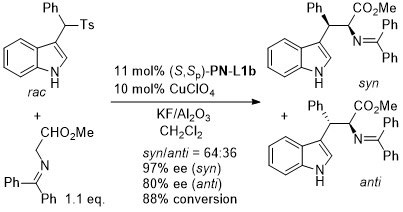
High anti selectivity and ee achieved with a complex obtained from AgCl/phosphoramidite.
Silver
Ag – 3 + 2 cycloaddition OL05-7-5055
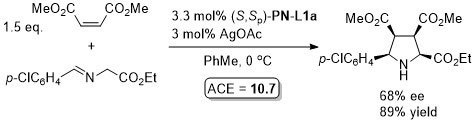
With the p-CF3C6H4 (instead of Ph) and benzyl (instead of i-Pr/t-Bu) analogue of (S,Sp)-PN-L1a/b these reaction result in >93% ee with a variety of aryl substituents (i.e. instead of p-ClC6H4).

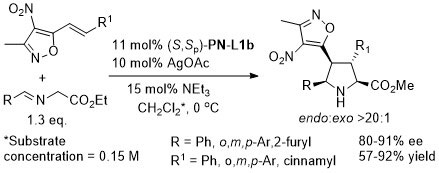
The description of the catalyst loading and reactions conditions employed differ in the manuscript and SI. SI conditions used in the above scheme. High exo selectivity (>20:1) and ee (82-93%) achieved with a phosphoramidite ligand (this S,R,R ligand will result in the enantiomeric series of products).
Ag – Conjugate addition/Friedel-Crafts cyclisation ASC18-360-2191
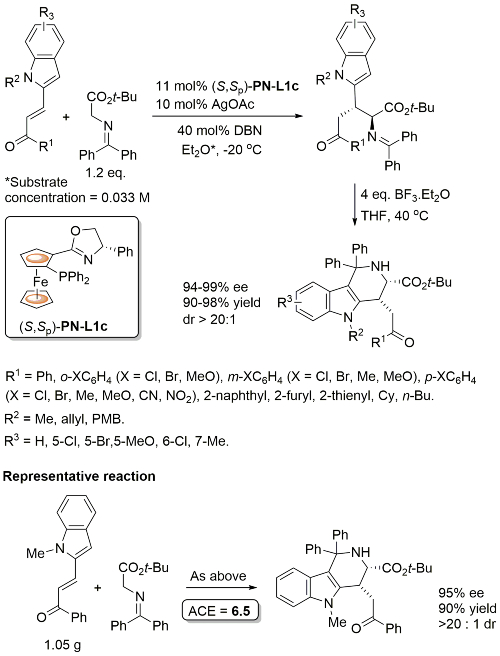
Conjugate addition chemistry closely related to similar Cu-based chemistry. See OL10-12-1080. In an intial ligand scoping study PN-L1a gave 76% ee vs. 87% with PN-L1c. Overall methodology also applied to a nitroalkene substrate (i.e. with COR1 replaced by NO2). The overall reaction is described as a [3+3] annulation. The alternative [3+2] cycloaddition pathway is essentially prevented (>20 : 1 selectivity) by the benzophenone derived azomethine ylide.
Nickel
Ni – Allylic alkylation Perkin100-2725
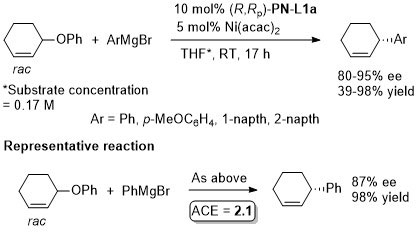
Ni – 2 + 2 + 2 cycloaddition JACS10-132-15836
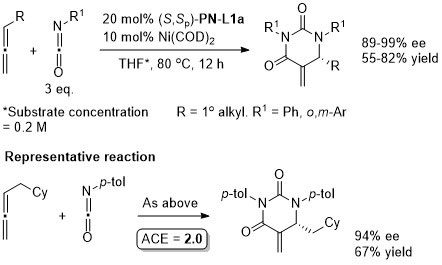
Ni – Denitrogenative annulation JACS10-132-54
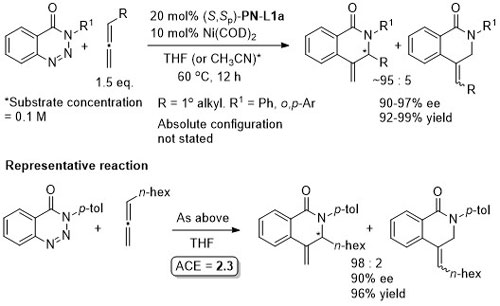
Absolute configuration not stated, but may be tentatively assigned as R by comparison to JACS10-132-15836.
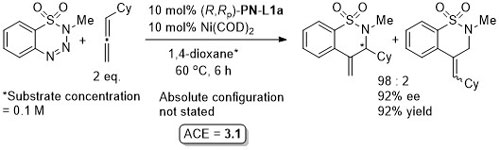
Absolute configuration not stated, but may be tentatively assigned as S by comparison to JACS10-132-15836. Reaction further exemplified using QUINAP.
Ni – Decarbonylative annulation OL11-13-1374
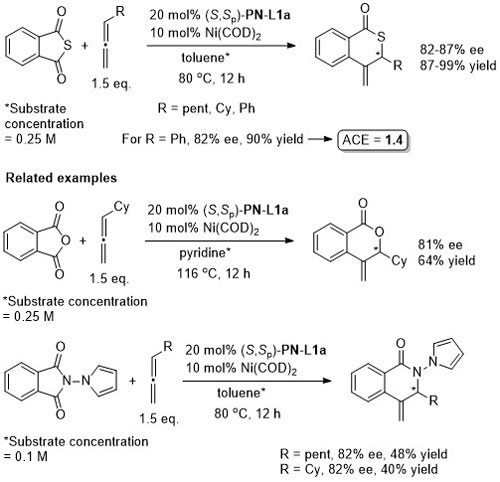
Absolute configuration not stated, but in all cases may be tentatively assigned as R by comparison to JACS10-132-15836.
Ni – Intramolecular alkene arylcyanation Synlett10-1709
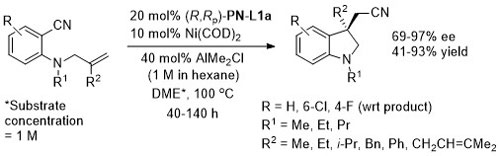
Specific example JACS08-130-12874
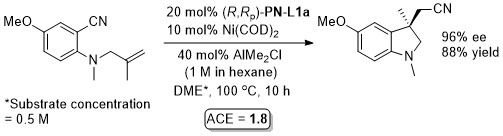
Ni – Reductive alkene-diarylation/cyclisation JACS18-140-12364
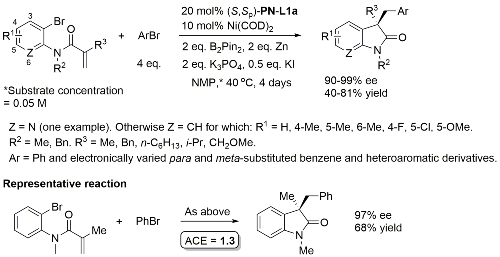
Ni – Diazabicycle desymmetrisation JACS17-139-15656
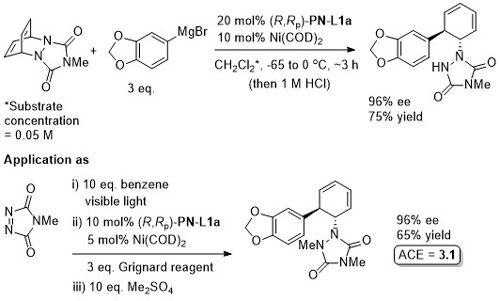
Intermediate in the synthesis of (+)-pancratistatin.
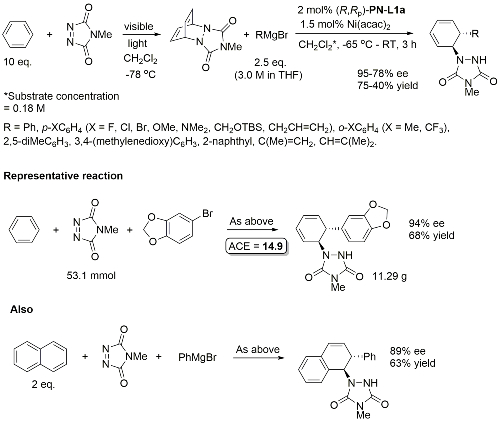
Yields quoted are for the overall process (photochemical cycloaddition and Grignard addition). Run as a one-pot procedure. Only one example with naphthylene as substrate (as shown).