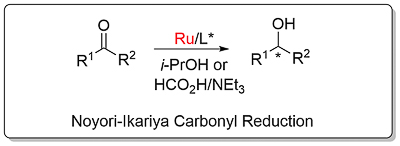
Noyori-Ikariya Carbonyl Reduction [Ru]
CBS Carbonyl Reduction [B]
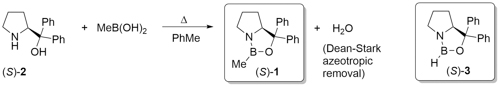
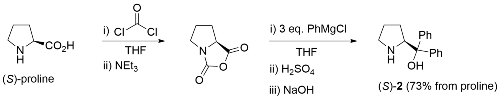
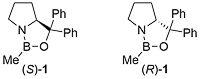
The CBS catalyst (S)-1 (CBS from its discovers: Corey, Bakshi and Shibata) is obtained from the reaction of amino alcohol (S)-2 and methylboronic acid with removal of water [JACS87-109-7925]. The B-methylated catalyst 1 is easier to isolate, gives higher ee values, and is more stable (it may be transferred in air) than the earlier B-H version (S)-3 [JACS87-109-5551]. Review = [Angew98-37-1986].
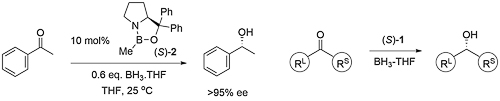
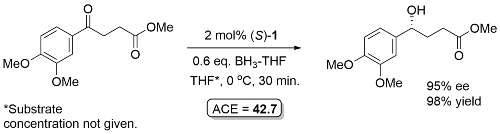
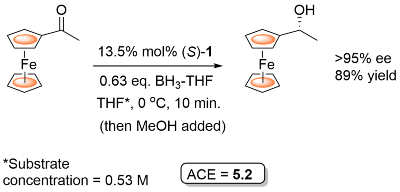
Ketone reduction reactions with (S)-1 as catalyst typically employ a 10 mol% loading with 0.6-1.0 eq. of BH3..THF as the reductant in THF at 0 oC or room temperature. A substoichiometric quantity of reductant BH3..THF may be used as up to two hydrogens are transferred. Reductant BH3..SMe2 may also be used. The reduction of acetophenone is representative (Scheme below). The secondary alcohol product is obtained after the addition of a proton source such as MeOH. To a good first approximation a simple mnemonic based on the relative size [RL = large (typically aryl), RS = small (typically alkyl)] of the ketone substituents predicts the configuration of the product alcohol. However it is not size operating through a disfavouring steric effect that principally controls enantioselectivity (see below).
The catalytic cycle involves the following steps: Step 1. Incorporation of BH3 = results in increased hydride donation ability and an increase in Lewis acidity of the catalyst boron. Step 2. Coordination of ketone substrate – activation as an electrophile. Step 3. Hydride transfer and asymmetric reduction. Step 4. Product release – regeneration of catalyst.

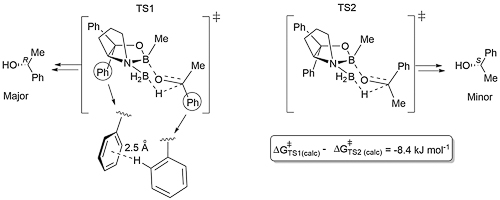
For step 3 calculations reveal a chair-like transition state structure with TS1 favoured over TS2, mainly as a result of a stabilising σ-π London dispersion interaction between the phenyl group of the substrate (C-H σ) and a phenyl group (π) of the catalyst [Angew21-60-4823]. The difference in diastereomeric transition states ΔΔG‡ = 8.4 kJ mol-1 (at 275 K) corresponds to an ee value of 95%, in excellent agreement with experiment. The polarizability per volume (α/v) of the R substituent of RCOMe increases in the order Et < n-Pr < t-Bu < Cy < Ph, and there is a reasonable correlation between α/v and ee. In a competition reduction experiment between t-BuCOMe and n-PrCOMe [with (S)-5 as catalyst, see below], in addition to the former resulting in a higher ee, it was also reduced at a faster rate. It was concluded that is a result of a stabilising LD interaction in the transition state for the reduction of the more sterically demanding substrate.
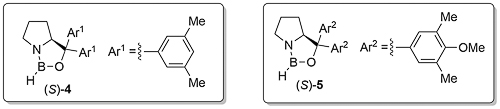
This interaction is increased where the catalyst phenyl group is replaced by a 3,5-dimethylphenyl or a 3,5-dimethylphenyl-4-methoxy group to give catalysts 4 and 5, and these can give significantly improved ee values with more challenging dialkylketone substrates.
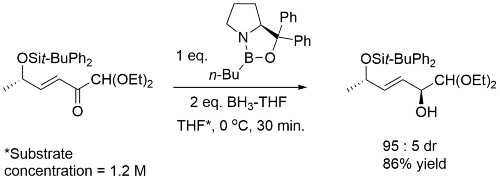
Examples
With corresponding (R)-proline derived catalyst the corresponding diastereoisomer predominated with dr = 97 : 3 (i.e. essentially complete catalyst control). See TL94-35-6681.
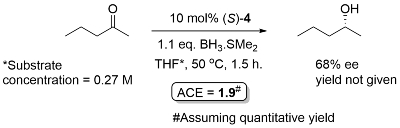
74% ee with 5, 64% ee with 3. Reaction quenched with 0.5 M citric acid solution (7.5 eq.).
Catalyst Precursor Synthesis. Readily synthesised from proline [JOC91-56-751]
Catalyst Availability
Precursor to 1 and 3
Catalyst 1
Aldrich (S) (R) Strem (S) (R) TCI (S) (R)
Precursor to 4

Aldrich (S) Key Organics (R) [250723]
Noyori-Ikariya Carbonyl Reduction [Ru]
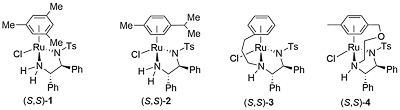
Ruthenium complex 1 was first reported by Noyori from the reaction of [RuCl2(η6-mesitylene)]2 and (1S,2S)-N-(p-toluenesulfonyl)-1,2-diphenylethylenediamine [(S,S)-TsDPEN)]. This was initially employed as a catalyst for asymmetric transfer hydrogenation of ketones employing isopropanol as the hydrogen source, but this reaction is limited by the reversibility of this process requiring a low substrate concentration of 0.1 M, and an avoidance of prolonged reaction times to prevent the erosion of product ee [JACS95-117-7562]. In contrast, the use of 5 : 2 formic acid-triethylamine azeotrope as solvent and hydrogen source results in an irreversible reaction and a more practical method of ketone reduction that may be run with a substrate concentration of 2 M or greater [JACS96-118-2521]. Reactivity as a function of the η6-arene substituent decreases in the order benzene > mesitylene (1) ≈ p-cymene (2) > hexamethylbenzene, and complexes 1 and 2 give higher ee than the corresponding benzene complex.
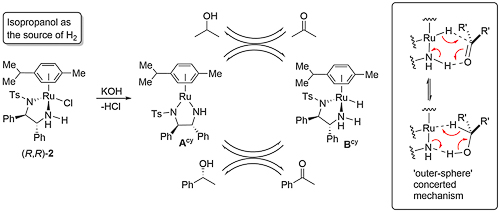
The true catalyst derived from 2 (where complex 2 is a representative example) is the Ru(II) square-planar 16-electron complex Acy (cy = p-cymene) resulting from elimination of HCl. This purple complex may be isolated [Angew97-36-285]. Reversible abstraction of dihydrogen from isopropanol results in the dihydride complex Bcy which reacts reversibly with a prochiral ketone to give the chiral alcohol product. Simultaneous hydrogen transfer is proposed via a concerted six-membered transition state in an ‘outer-sphere’ mechanism [JACS00-122-1466], although calculations incorporating solvation have identified an ion-pair intermediate of a step-wise mechanism (hydride addition and then proton transfer) [JACS13-135-2604].
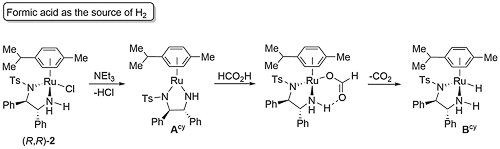
With formic acid/triethylamine Acy is also formed by abstraction of HCl, followed by generation of a formate complex which undergoes loss of CO2 to generate dihydride Bcy [ASC04-346-37]. Although this CO2 reaction is reversible, under the reaction conditions removal of CO2 makes this step essentially irreversible (the addition of CO2 was performed at a pressure of 10 atm.)
The dihydrogen intermediate formed from (R,R)-Ames (mes = mesitylene) can be either the (R,R,SRu) or (R,R,RRu) diastereoisomer with ruthenium as a stereogenic centre containing four substituents in a pseudo-tetrahedral arrangement. Calculations with isopropanol as hydrogen source reveal that complex (R,R,SRu)-Bmes is both kinetically and thermodymanically prefered compared to (R,R,RRu)-Bmes [ACSCat21-11-13649]. Furthermore, (R,R,SRu)-Bmes is matched and (R,R,RRu)-Bmes mismatched with respect to enantioselectivity and rate of of acetophenone reduction, such that only the former need be considered to account for product enantioselectivity (eecalc = 93% is in good agreement with experiment).
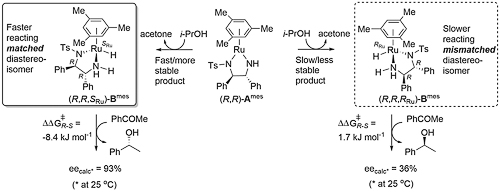
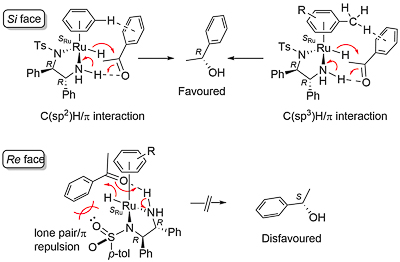
The origin of enantioselection was initially ascribed to resulting primarily from acetophenone (as representative substrate) interaction with the η6-arene ligand, with DFT calculations identifying a transition state stabilising C(sp2)-H/π interaction with η6-benzene, or a stabilising C(sp3)-H/π interaction with η6-hexamethylbenzene [Angew01-40-2818]. Later studies suggested this was not the sole basis of the selectivity, and in particular identified a repulsive S=O oxygen lone pair/π interaction disfavoring hydrogen addition to the alternative prochiral face of acetophenone [JACS13-135-2604].
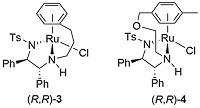
A number of complexes closely related to 1 and 2 have been reported of which the most significant is the N-Ar tethered complex 3 reported by Wills. This is more active than 1 and 2 for asymmetric transfer hydrogenation of ketones in 5 : 2 formic acid/triethylamine, has significantly improved longevity, and in many cases gives improved enantioselectivity. It may be used with a catalyst loading as low as 0.01 mol% [JACS05-127-7318]. A closely related tethered catalyst 4 was introduced later by Ikariya [JACS11-133-14960] with evidence to suggest that the oxygen in the tether further increases the catalytic activity.
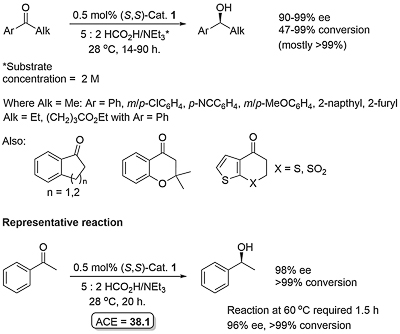
Examples

This method is not applicable for the generation of alcohols such as 1-phenylethanols containing electron-donating substituents (e.g. p-MeOC6H4) because the significantly faster rate of the reverse reaction results in significant erosion of product ee. This has been exploited for the kinetic resolution of these alcohols using 1 or 2 as the catalyst in acetone [Angew97-36-288].
With Alk = Me and Ar = 1-naphthyl ee = 83%. No other examples with an ortho-substituent.
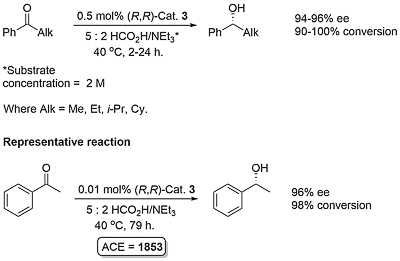
For acetophenone the same ee (96%) was obtained at 40 oC as at 28 oC. Cyclohexyl methyl ketone was reduced using (R,R)-3 to to give the corresponding (S)-alcohol with a 69% ee (i.e. with the opposite facial selectivity of acetophenone).
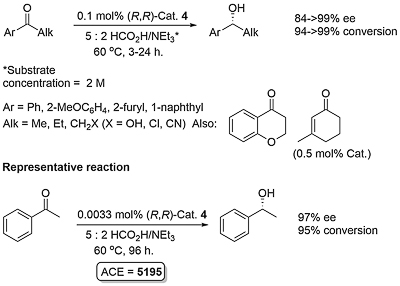
Cat. 4 abbreviated to Ts-DENEB. Reaction time with 0.1 mol% loading typically 5 h.
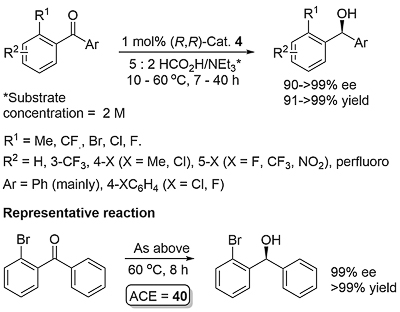
Selectivity explained by the R1 ortho substituent forcing the corresponding aryl group out-of-plane with C=O, such that only the in-plane aryl group (typically Ph) participates in CH-π interaction. High ee values (>90%) were also obtained for a number of diarylketones Ar1COAr2 containing one or two meta-NO2 or F substituents on one aryl group (the other typically Ph), the argument here being that the electron-poor aryl group switches off the CH-π interaction. However, this electronic basis for selection in itself appears unable to completely control selectivity. For example, with Ar1 = p-NO2C6H4 and Ar2 = p-MeOC6H4 an ee of 79% was obtained (product = R). Significantly, benzoylferrocene was reduced slowly in 90% ee (product = S). As acetylferrocene does not appear to be a suitable substrate for this reduction method, this outcome further highlights the significance of CH-π interaction (which does not appear to operate with ferrocene).
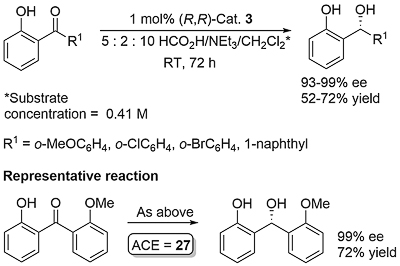
Reactions carried out in CH2Cl2 to aid solubility. Significantly lower ee if the substituent on R1 is not ortho (as for the ortho-substituted examples in JACS16-138-10084, the substituent forces the aryl group to be out-of-plane with C=O, such that the o-phenol group, for which OH is too small to prevent planarity (OH—O=C hydrogen bonding), participates in CH-π interaction). With R1 = Ph the same product configuration results but in 80% ee due to electronic differentiation between the two aryl rings. These two influences work together to give high ee. For the representative reaction use of 2 gave 73% ee, further highlighting the improved selectivity offered by tethered 3.
Catalyst availability
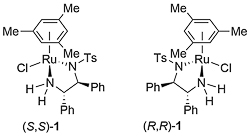
Aldrich (S,S) (R,R) Strem (S,S) (R,R) TCI (S,S) (R,R)
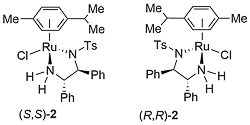
Aldrich (S,S) (R,R) Strem (S,S) (R,R) TCI (S,S) (R,R)
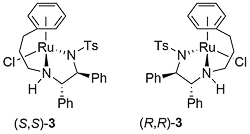
Aldrich (S,S) (R,R) Strem (S,S) (R,R)
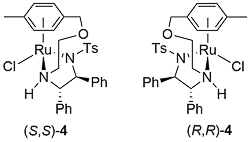
Aldrich (S,S) (R,R) Strem (S,S) (R,R) TCI (S,S) (R,R) [200823]