Hydrosilylation
Hydrogenation
Noyori-Ikariya Imine Reduction [Ru] [HCO2H as hydrogen source]
Buchwald Hydrosilylation
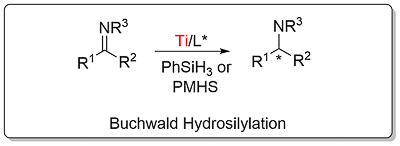
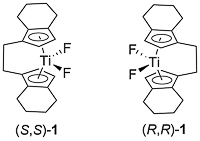
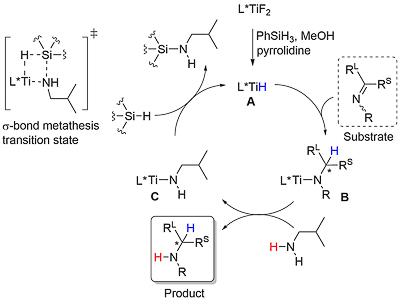
This method for the asymmetric hydrosilylation of imines was first reported by Buchwald in 1996 [JACS96-118-6784]. It builds on earlier work by the same group on the use of a Brintzinger ansa-titanocene [JOMC82-232-233] derived catalyst for asymmetric hydrogenation [JACS94-116-8952]. In contrast to hydrogenation reactions which require a high hydrogen pressure (80—500 psi), hydrosilylation reactions typically require heating to 60 oC under an atmosphere of argon. The first series of substrates were N-methyl imines as the reaction was found to be very sensitive to the size of the nitrogen substituent. This was solved by the adding an excess of iso-butylamine slowly by syringe pump over the course of the reaction (several hours). Activation of (air-stable) 1 is achieved by heating with PhSiH3, piperidine and methanol (4 equivalents of each with respect to the catalyst). This is thought to generate in situ titanium(III) hydride A. Imine insertion into the Ti-H bond gives amido complex B which exchanges with iso-butylamine to give the reduced product and new amido complex C. This less hindered complex undergoes reaction with the stoichiometric silane reducing agent by sigma-bond metathesis to regenerate A. In the absence of iso-butylamine the more sterically encumbered complex B must participate in the step to regenerate A (only viable where R = Me). Slow addition of iso-butylamine is required as if present in high concentration this generates H2 on reaction with A. A further significant advantage of employing iso-butylamine is the viability of using PMHS as silane, although PhSiH3 is still used for the generation of A.
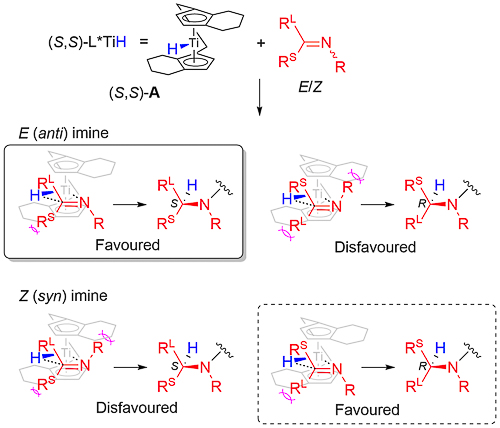
Enantioselectivity resulting from imine insertion into the Ti-H bond of A can be explained as follows using (S,S)-A derived from (S,S)-1. For an imine of E configuration the favoured lower energy pathway leading to the S product minimises interaction of the RL and R substituents with the hydrocarbon framework of the catalyst. For an imine of Z configuration the same argument with the RS and R substituents favours the pathway to the R enantiomer. Catalysed hydrosilyation reactions with low E/Z isomer ratios can lead the product expected from the E pathway in high ee suggesting that imine isomer interconversion occurs under the reaction conditions. This contrasts with asymmetric hydrogenation which also proceeds via A and for which the product ee is much more sensitive to the E/Z ratio [JACS94-116-8952]. The favoured pathway for the E isomer has a RS/framework steric clash in contrast to the Z isomer for which there is a corresponding RL/framework clash.
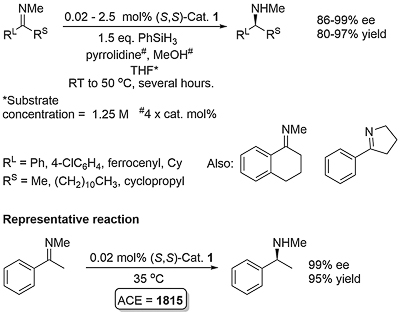
Product obtained after acidic workup to hydrolyse the initially formed silylated amine.
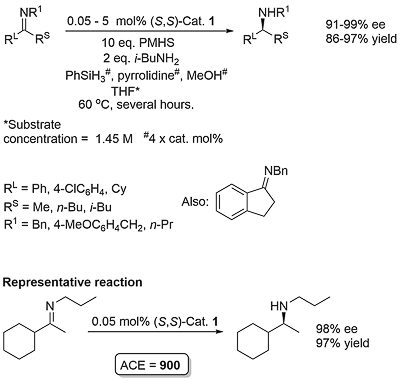
Product obtained after acidic workup to hydrolyse the initially formed silylated amine. The reaction is not dependent on a high E/Z imine isomer ratio (in some cases as low as approx. 2: 1). Slow addition (syringe pump) of iso-butylamine required. Substrate with RL = n-hexyl, and RS = Me (R1 = Bn) gave an e.e. of 69%.
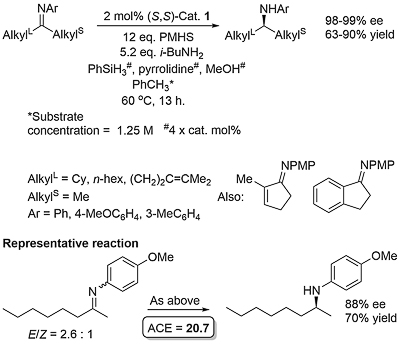
Product obtained after acidic workup to hydrolyse the initially formed silylated amine. Slow addition (syringe pump) of iso-butylamine required (over the 13 h reaction time). The N-aryl substrates are less reactive than N-benzyl substrates and thus a 2 mol% catalyst loading was employed. Except for the 1-indanone derived imine, imines derived from aromatic/alkyl ketones gave low ee values.
Catalyst availability
Aldrich (S,S)
Noyori-Ikariya Imine Reduction (see here for carbonyl reduction)
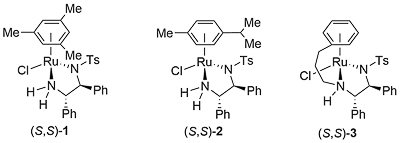
Following the first report by Noyori (JACS96-118-4916), application has focused mainly on the reduction of dihydroisoquinolines (DHIQs) and related substrates. Examples of acyclic imine reduction are limited, in part because of the challenges presented of E/Z isomerisation. Calculations with the N-benzyimine of acetophenone as substrate point to the minor Z imine isomer resulting in more of the opposite product enantiomer to that formed from the major E imine isomer (Orgmet12-31-6496). A number of ammonia derived imines (i.e. no E/Z issue for a protonated substrate) containing a 2-hydroxyphenyl substituent work well (OL14-16-2310, OL20-22-3717). The sense of asymmetric reduction with acyclic imines is the same as the corresponding ketones. Significantly, endocyclic C=N bonds such as in a DHIQ substrate are reduced with the opposite selectivity, and a modelling study has proposed an explanation (Orgmet11-30-4822).
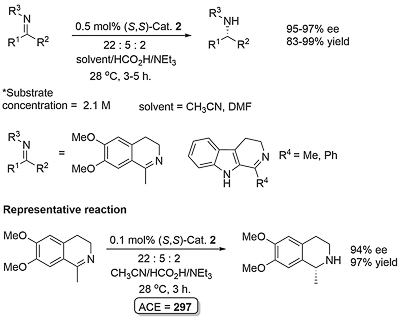
Reaction very slow in neat formic acid/triethylamine. Under the conditions used ketimine > 1000 times more reactive than the corresponding ketone. The N-benzylimine of acetophenone resulted in the (S)-amine product in 77% ee with an (S,S)-catalyst related to 1 and 2 (η6-arene = benzene and NSO2Ar with Ar = 2,4,6-Me3C6H2). The lower ee values with this acyclic imine are acscribed to syn - anti isomerisation.
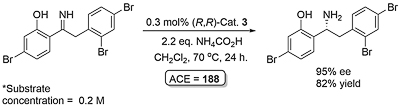
Imine formed form precursor ketone with dry ammonia in methanol (7 N). Vessel pressure of 39 psi during the reaction. Product in 99% ee after recrystallisation – an intermediate for the synthesis of MK-8742 (for treatment of hepatitis C).
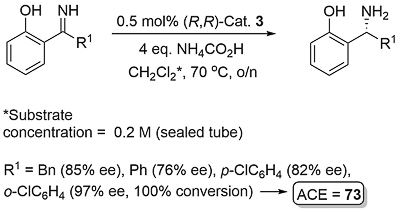
With R1 = Bn product ee = 32% with Cat. 2. Reduction of the imine derived from o-hydroxyacetophenone was not successful.
Ir/ligand/H2
Ir/Josiphos catalysts have been applied to imine hydrogenation, notably for the manufacture of (S)-metolachlor. A useful (open access) review on chiral amine synthesis by asymmetric hydrogenation (and therefore including imine hydrogenation) is CR22-122-269. A Phosferrox ligand has also been applied to this reaction.
240723